Structural Biology as an infrastructure of Biotechnology R & D
Japan Tobacco Inc. Masashi MIYANO
Structural Biology at the atomic resolution is crucial for the biotechnology R & D now and in future. The wealth of the biomacromolecular structure allows us to apply for new era of biotechnology in conjunction with the manipulation and production technology of the biomacromolecule like recombinant DNA. DNA synthesis, monoclonal anti-body and PCR technic as well as fermentation and purification process, however we still need to develop the technology for the structure biology as a broad interdisciplinary field for the experimental structure determination by X-ray crystallography, NMR, and electron microscopy. The studies on proteins in any aspects of each specific protein are only way to expand the experimental structural studies, while many genom projects are going on successfully. It will be needed a scale-up facilities for the preparing the sample for the structural biologyas well.
Now, under the tight cooperation with the present data-bases such as Brookhaven PDB and Cambridge Structure Databese the contribution of the structural data-base can be re-boosted in Japan like GenomNet and DDBJ in the genom data-base as an integrated center for the theoretical structural biology as well as the collection, verification and distribution of the indispensable structure data. I believe such data-base should be one of most important infrastructure for the future biotechnology R & D in structural biology.
なぜいま構造生物か?
1970年代後半から予想をはるかに超えて発展してきた遺伝子工学の技術をべ ースにしてきた分子生物学、タンパク質工学、抗体工学、進化工学といった技術は 生体由来の分子をそのまま相手にする工学といえるだろう。これまでの工学は、機 械は筋肉の延長として、マイクロエレクトロニクスは神経の働きの代替えとして、 生体が実際に使っているものとは全く違う素材と物理原理に基づいて生物界に見ら れる機能をミミックしてきた。ところが、この広い意味でのバイオテクノロジーは 生体の構成物質を、生体内で働いているのと同じ作用機構を利用して行うという意 味で全くこれまでの工学とその実において異なっている。そして、生体中で機能を 担っているタンパク質、RNA,DNAなどの分子の原子構造そのものが機能その ものと不可分であることが、ワトソン-クリックによるDNAの2重らせんモデル 以来、今ではあたりまえのことと理解されている。
産業利用の上でこの分子の原子構造に実用的意味を持ちうるのは化学構造と合成 化学のように手で扱い作り出す技術が存在するときである。バイオテクノロジーに おいて思い通りにつくったり変えたりする技術とは、遺伝子組み替え、PCR, D NA合成技術であり、モノクローナル抗体技術であり、そして生産するための発酵 培養技術と精製技術である。
このタンパク質の工学的基礎となっているのはまさにワトソン-クリックによる セントラルドグマであり、情報と機能の担い手がそれぞれDNA、タンパク質と、 物質的基礎を全く別にしている。(ここでは話を簡単にする意味からと、現在のと ころ実用上の主要な対象はタンパク質であることでもあり、タンパク質が生体機能 の担い手であるとするとして議論を進める。ただし、RNAは情報、機能両者の間 に介在しかつ両方の機能を持ちうる特殊な存在ではある。)そしてこのセントラル ドグマが現代のマイクロエレクトロニクス化した生産設備のソフトウェアのように 情報担い手であるDNA配列の書き換えをしてやれば、機能の担い手を自由に変え られることを保証している。そして今ではこの塩基配列のテープをコピーしたり、 切ったり張ったりするための多くの生物由来のものを加えて道具はかなりそろって いる。
どんな情報がこれまでに蓄積されたか?
これまでのDNAの配列はそしてもとになる配列はGenBankやEMBLの遺伝子デ ータベースを運用しているコンピューターデスクにはすでに知られた億を越えるD NA配列に相当する何ギガビットの磁気記憶の列が記録されている。そして、PIR、 PRF、Swiss Protなどのタンパク質データベースには2000万残基に上るアミノ酸配 列が登録され、PDBには100万を越えるアミノ酸配列と3500を越えるタンパク質 の原子座標が控えている(GenomeNet WWW server 1995)。
タンパク質遺伝子の数と立体構造の数。
C. Chothia(1992)はいろいろな生物の遺伝子解析の結果とPDBに1991年まで に登録された1000あまりのタンパク質の原子座標から似たような立体構造をもつ タンパク質のfo1ding motifは高々1,000しかこの地上の生物には存在しないと推定 した。この推定値はさておき、可能なアミノ酸配列の組み合わせのうち、実際に機 能しているタンパク質の立体構造の折れ畳み構造のパターンの存在はきわめてまば らなものであり、そのうえ似たものはたくさんあるだろうというのは、上記のデー タベースの結果からもっともらしく感じる。1992年に1000あまりのPDB の座標データから254ファミリーで83種のfo1dsがあるされたものが、199 5年の1月リリースのPDBの3000を越すタンパク質の原子座標は、せいぜい 400の異なったファミリーに属しており、そのfo1ding motifは150を越えない と推定されている(T. J. Hubbart and A. M. Lesk 1995)。これが真実ならば、宇宙で の星の分布の偏りなどというレベルをはるかに超えている。これが進化的制限によ るのかアミノ酸の折れ畳みの物理化学的原理によるのかは重要なopen quentionか もしれないが、このギャップがあればあるだけ解かれた構造のデータベースの重要 性は新たなタンパク質の構造を解くという観点からだけでもますます重要になる。
今のところ実験的にしか確からしいタンパク質の構造モデルはできない。
DNAの配列として決められた1次元的アミノ酸配列が機能の担い手として働 くためには原理的には予定調和的に自ら折り畳まれて触媒分子、機械分子、信号変 換分子、運搬分子、構築分子として機能を果たすための正確な立体構造をとるよう に空間配置をもつことが、あるものはシャペロン助けを必要とし、あるものは翻訳 後修飾などがされることがあっても、原理としては正しいと信じられている。この 自己組織化.としての折り畳まれ方は、現在のところアミノ酸配列だけから予測こと は困難な課題である。タンパク質の機能構造とそれを構成しているアミノ酸の1次 配列の間に経験的予測はあるにしてもそのα-ヘリックス、β-シートといった単 純な2次構造予測でさえも信頼できる演繹的方法があるとはいえない。現在のとこ ろ、実験的に正しく立体構造を決めなければ分子設計に耐えるだけの十分に信頼の 置けるタンパク質を構成する原子の空間配置を決められない。だから、まだまだど んなに手間がかかろうとも働く分子のかたちはX線結晶解析、NMR、電子顕微鏡 など原子レベルでの構造解析が可能な実験的方法で決めていく必要がある。
構造解析の実験には最高品質の大量のタンパク質が必要である。
国際的なゲノムプロジェクトの進展のなかで新たに発見された大量の新規タンパ ク質の遺伝子機能の解析を進めるためには、生体の働く分子と同じかたちをしたタ ンパク質の生産が不可欠である。原子構造レベルでのタンパク質構造決定はいずれ の方法によれそのタンパク質の機能構造解析のひとつの到達点であることから削る とおり、対象となっている個々のタンパク質の研究そのものが進まなければ構造解 析をする環境も実際に解析すべきタンパク質標品も提供されることは難しい。一般 にDNAの取り扱い技術に比べ、働くかたちをした分子としては個々のタンパク質 によって、物性、機能ともに大きな違いのあるタンパク質についてはそれぞれのタ ンパク質としてしかその安定性、精製法などが議論できないなかではタンパク質研 究が進むことが構造解析にふさわしい標品の提供を進めるだろう。
構造解析のための標品タンパク質生産。
X線結晶解析には当然ながら結晶が必要であり、後者にも前者の結晶化用標品同 様大量のタンパク質が必要である。結晶化のためのタンパク質標品はその立体的構 造を含めて均質性が必要であり、私たちは少なくともネイティブホリアクリルアミ トゲル電気泳動的(NATIVE-PAGE)に均質であることを最低限としており、しばし ばSDS-PAGEで均質に見えるものでも多量体が存在したり、染色したバンド に広がりが見られる。このほかには、等電点電気泳動法がより感度がいいが NATIVE-PAGE同様万能というわけにはいかない。これ以外には光散乱を用いてそ の水溶液中でのhydrodynamicな性質を決定して平均の分子量とともにその分子形 状の均質性を結晶化の条件に近いところで測定することで結晶化の可能性の可否を 判定できるとの説もある。いずれにしても、タンパク質結晶解析に用いるタンパク 質標品はそのとき得られる最高の標品であることが当たり前といえる。結晶解析の ために空気酸化されて不安定である遊離型のシステイン残基をアラニン、セリンな どに置換する、また、膜結合部位を切り取ってしまったり、重原子サイトを導入す ることは決して珍しくはない。またNMRは比較的微量の不純物は問題にしなが、 必要なタンパク質標品の量は結晶解析以上である。さらに、ある程度の大きさの分 子量を持つタンパク質であれば、安定同位体ラベルをした標品が信号の重なりをさ けるために必須となってくる。このようにタンパク質の原子レベルでの立体構造解 析には最高レベルのタンパク質の抽出精製が必須である。このためにはタンパク質 の生化学的、物理化学的研究の高いレベルが要請される。
これと同時にタンパク質の100mg単位の発現精製には細胞、菌の発酵培養が 装置ともどもファシリティーとして必要である。それは、試験管レベルでの発現方 法は1リットルまして10リットルをこす培養はそのままではうまくいかないのが 普通であるからだ。
コンピューターによる信頼できる構造モデル構築は大きな期待である。
これまでのところは実験的にしか、なかなか信頼のおける構造モデルができなか たとしてもコンピューターによるモデリング技術は進歩してきており、特に、タン パク質のfoldingmotifの数の推定のように似たようなかたちをしたタンパク質の数 がきわめて限られていたとして現在のこのぺ一スで既知の構造のタンパク質が増え ていけば、今後のコンピューターモデリング技術の発展の与える寄与は広範なもの になる。いまのモデリング技術でも、よく似たタンパク質の立体構造が知られてい ればだいたいの立体構造をhomology mode1ingの手法によって推定できる。
homology mode1ingをする前にはどのfolding motif のタンパク質ファミリーに属する かアミノ酸配列から確実に決められなければならない。タンパク質分解酵素でもグ ロビンタンパク質ファミリーなどアミノ酸配列ではほとんど類似性が認められない ものでも立体構造がよく似ていたということはすでに珍しくない。20%以下さら には5%以下のアミノ酸相同性の同じfolding motifをもつタンパク質をいかに知る かも残された課題である。組み上げたモデルの精度という点でもさらにまだまだ克 服すべき課題が多い。現在の標準的な力場を用いた分子動力学を用いた計算では、 これまで水を入れない計算では発散してしまい、現在の分子動力学計算では系に水 をいれることが必須となっている。実際に水を入れた計算については、十分な時間 とは考えられてはいない100ピコ秒程度計算したものでも小さいタンパク質を用い て両手に足りないほどの報告があるだけだ。さらにコンピューター能力の飛躍的進 歩と信頼性の高い力場の開発など計算法の発展の両方に大きな進展が必要である。 これによって精度よい類似タンパク質のモデル構築が可能となれば、利用できる構 造情報が飛躍的に豊かになる。
コンピューターモデリングの成功には理論そのものの発展が必要である。
標的タンパク質の構造をべ一スとした阻害剤開発による、医薬農薬の開発( Protein Structure-based Drug Design)のためには、リガンドータンパク質複合体の 解析のために分子間相互作用の見積もりが不可欠であるが、残念ながら実感として まだまだタンパク質の立体構造中での水の寄与を含む疎水的相互作用、水素結合そ して速達力であるイオン相互作用の寄与の見積もりは難しい。例えば、M. Pertzに よって提唱されたベンゼン環のπ電子に対する水素結合は多くの具体例が構造決定 されたタンパク質の構造中に有為に存在することが確かめられたが、計算化学の手 法では ab initio 計算をする以外のエネルギー的寄与の見積もり法を私は知らない。また、分子動力学により自由エネルギー摂動計算(free-energy perturbation method)による相互作用の見積もりはより精度の高い力場開発などの計算法、そしてコンピ ューターの能力ともに大きな課題が残されている(A. E. Mark and W. F. Gunsteren 1995)。
タンパク質の原子レベルでの構造解析手段。
原子レベルでのタンパク質分子の主たる構造決定方法はこれまでも将来も当分は X線結晶構造解析が中心である。まさに飛躍的発展をしているNMRはX線結晶解 析と互いに相補う関係にあって、良きライバルであり両者がパートナーシップで研 究協力できることが理想的である。このほかにも、電子線回折による構造解析も視 野にはいいりつつあるかもしれない。ちなみにMacromolecular Structure1995に よれば1994年中に新規に決められたタンパク質、核酸はX線結晶解析で352、 NMRで100、電子顕微鏡で2つであったという(W. A. Hendrickson and K. Wuthrich1995)。それぞれに長所短所があり、これら以外の方法であって可能性 があればそれを生かして立体構造情報を豊かにして次の飛躍のステップになること を期待する。最近のJ. P. Gluskerによる Acta Crystall. section D の編集はその視野の広さに高い見識を感じるとともに構造生物の一翼を担うタンパク質X線結晶構造 解析のあるべき一つの方向を明確に指し示していると思う。
構造解析のためのインフラとしてのタンパク質立体構造データベース。
これまでのタンパク質などの構造の解かれた生体巨大分子の原子座標を集めてい るPDBは、構造解析自身にとっても不可欠である。タンパク質X線結晶構造解析 の現在の主要な解析法である分子置換のための初期モデルとして私たち自身、きわ めて多くを負っている。分子置換法ではその構造の半分以上はこの初期モデル自身 により構造決定されたといっても言い過ぎではない。そして、PDBから派生した 部分構造データベースは構造モデリングの重要な構成要素となりつつある。
もう一つのバイオテクノロジーにとって重要な構造データベースCambridge Structure Database(CSD)は、R.Huberら(1993)によりタンパク質の精密化のため の理想的なアミノ酸残基の原子配置を改訂するのに使われた。現在の、タンパク質 結晶構造解析の結果は、精密化して初めて正確さと精度において完成とみなされる ようになっている。そして、精密化の結果からX_PLOR,PROLSQ,TNTなど、どの 精密化プログラムを用いたか原子座標の統計値から確実に推定できるという(J. P. Priestle 1995)。つまり、どのように精密化するかが結果にそのまま影響する。
また、データベースの機能のなかには当然新規構造モデルの原子座標、構造因子 データの収集が重要な部分である。構造データの収集にはデータベースの信頼性を 維持するために、提出された構造の正確さと精度を確認しなければならない。そし て収集したデータの効果的利用法の開発はこうしたデータベースの価値をより一般 的に広める。すでに、3次元的にタンパク質モデルをリアルタイムで「鑑賞する」 ことはGlaxoのR. Sayle(1995)によるRasMolなどと強力になったパソコン使えば誰 でも簡単に気軽にできるようになり、PDBデータをその3次元の構造として楽し める。構造化学は実験的蓄積が新たな理論を必要とするようになり、理論の進歩が 新たな実験的構造解析を押し進めることは歴史が示すところであり、特に非共有結 合に関する発展のためにも理論的構造論までが構造生物の将来的な視野に入ってく ることを期待する。
構造データベースに一部の特に見識ある日本の研究者がPDBに早くから参画し てきたことは最近知ったばかりであるが、私見では、残念ながら構造データベース の利用に比べ維持開発に対する日本の実質的寄与はまだまだと感じており将来に対 する不安を禁じ得ない。1企業の1研究者として個人的にはこうして現在PDBな どが利用で'きることが今後も研究を進める上で必須であると感じており、これらの 構造データベースに可能になった時点でわずかな寄与とはいえ義務として積極的に 寄託することで自分たちの構造決定の批判を仰ぐという姿勢でやってきた。個人を 越えて、これからの世界の中での日本として、もっとも基礎的なインフラの一つと して既存のデータベースと連携をとって統合した構造データベースヘの参画をしっ かりした長期的展望にたって早急に考える時期であると信じる。
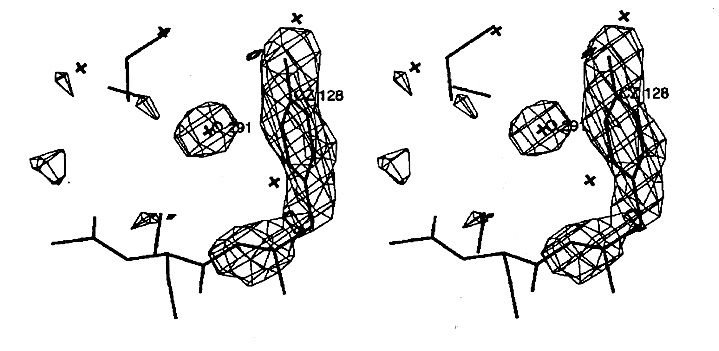
α-PAPの基質結合部位の溝にあるのTyr128のフェノール環のπ電子と水素結合している考えられると孤立した水分子H2O291のFo-Fc電子密度図。ベンゼン環平面の中心と水の酸素の距離は3.1ÅこのH20291はTyr128の骨格のアミド基の窒素とも水素結合距離(3.1Å)にある。
(PDB accesion number lAPA; H. Ago et al. 1994)