Structure of cytochrome c oxidase from Paracoccus denitrlficans at 2.8 A resolution.
Max-Planck-Institut fuer Biophysik So IWATA
The crystal structure of cytochrome c oxidase from Paracoccus denitaficans at 2.8 Å resolution is described. The crystallization of the membrane protein complex is acieved by co-crystallization with antibody Fv-fragment. The complex contains four subunits. Subunit I contains twelve tranmembrane helices and binds heme a and the heme a:3-copper B binuclear center where molecular oxygen is reduced to water. Two proton transfer pathways, one for protons consumed in water formation and one for 'proton pumping' could be identified. Possible mechanisms for proton pumping are discussed.
我々のパラコッカス由来のチトクローム酸化酵素の全構造[1]及びミトコンドリア由来の 酵素の金属部分の構造[2]が相次いで報告されたことにより,チトクローム酸化酵素の研 究は新局面を迎えた.
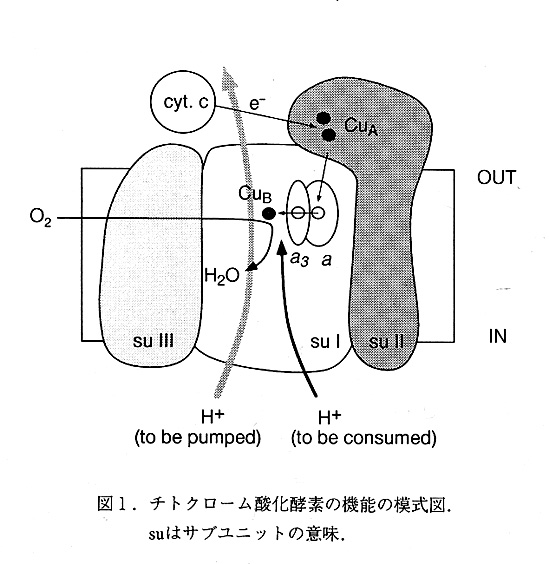
チトクローム酸化酵素は,細菌からミトコンドリアに至るまでその内膜に存在する膜蛋 白質複合体であり,酸素呼吸の呼吸鎖の最終酸化酵素である.この酵素はチトクロームC から分子状酸素への電子の伝達を触媒し,ヘムと銅原子を含む最終酸化酵素のスーパーフ アミリーに属している.酸素分子を二分子の水に還元するときに4個のプロトンを消費す るとともに,4個の別のプロトンを膜を横切って輸送する働きをもっていて,プロトンポ ンプとも呼ばれている(図1).このポンプによって生じた膜を隔てたプロトン濃度勾配 は,合成酵素などの,膜に結合したエネルギー変換システムによって,ATPなどのよ り生体に有効なエ不ルギーへと変換される.
我々は,チトクローム酸化酵素の構造研究の対象として,パラコッカス(Paracoccus denitrificans )由来の酵素を選択した.この酵素は4個のサブユニットからなるが,これま でに知られているすべてのチトクローム酸化酵素において,この内のサブユニットIから IIIが保存されている.また,このうちサブユニットI及びIIだけで酵素は完全に機能するこ とが知られており,それ以外のサブユニットの働きはよく知られていない.サブユニッ トIはプロトンポンプの本体であり,ヘムa及びヘムもと呼ばれる二つのヘムA分子と, Copper-B(CuB)と呼ばれる銅原子を含んでいる.ヘムもとCuBからなる"binuclar center"は分 子状酸素を水分子に還元する反応中心である.サブユニットIIはCopper-A(CuA)と呼ばれ る,二つの銅からなる金属中心を持っており,チトクロームcからの電子が最初に伝達さ れる部分である.
パラコッカスは"生きている ミトコンドリア"ともいわれ, と考又られる細菌とも密接に 関連していると思われる.パ ンドリアのものによく似てお アの酵素に対し70%程度の非 知られている.また,酵素の 機能解析において不可欠な変 異酵素の作成がパラコッカス を含む細菌のチトクローム酸 化酵素のみで可能である.我, 々は,このチトクローム酸化 酵素の機能の解析を行なうた めに,本酵素の結晶構造解析 を行なった.またこの酵素の結晶を得るために,抗体のFvフラグメントと本酵素の複合体 を形成し結晶化を行なうと言う新技術を開発した[3].本論文では,この抗体フラグメン トを使った膜蛋白質結晶化とチトクローム酸化酵素の構造と機能についてまとめた.構造の細部について,及びプロトンポンプの機能については原論文[1,3]をあたっていただければ幸いである.
抗体Fvフラグメントを用いたチトクローム酸化酵素の結晶化
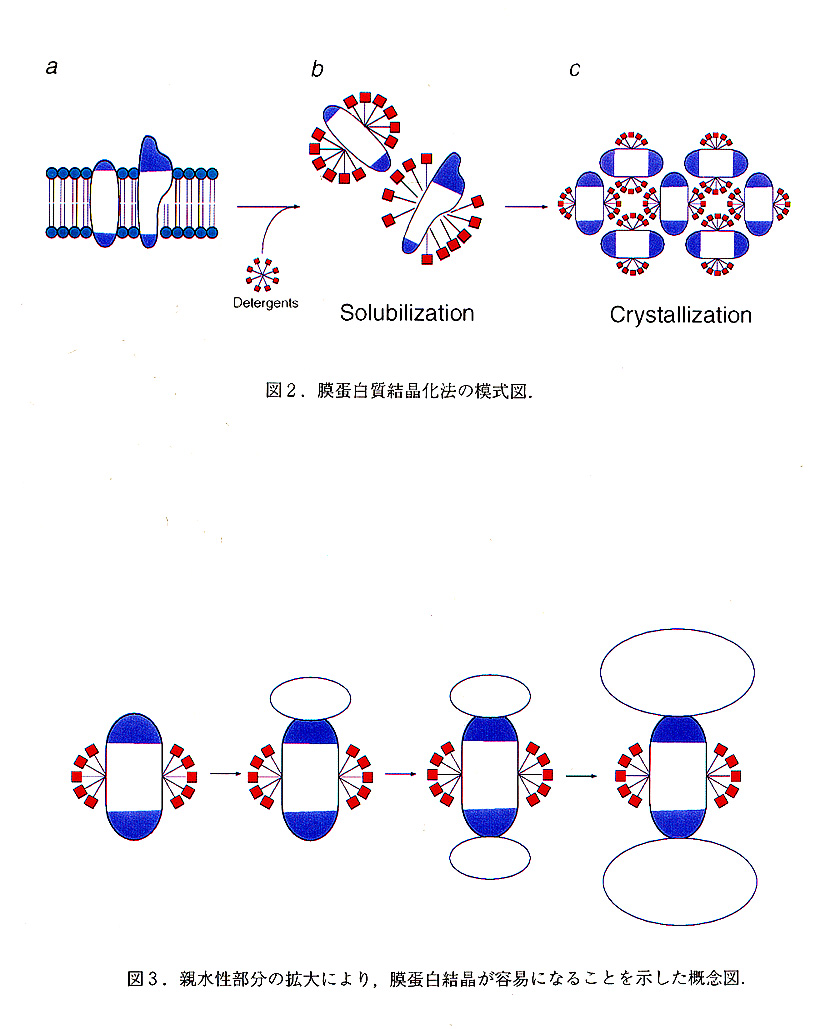
これまで,20年近くにわたって多くのグループが本酵素の結晶化に取り組んできたが, 良好な結晶は得られていなかった.本酵素に限らず多くの膜蛋白質の結晶化は,依然科学 と言うよりはむしろ芸術といえるほど難しい.図2に簡単に膜蛋白の結晶化についてまと めた.図2の一番左側に示したように,膜蛋白質は脂質二重膜の中に存在しており,膜に 埋もれた部分は疎水性,外に露出した部分は親水性である.膜蛋白質を結晶化するために は,まず膜蛋白質を界面活性剤で可溶化してやる必要がある.膜蛋白質を変性させずに可 溶化するために,マイルドデタージエントと呼ばれる一連の非イオン性または両イオン性 界面活性剤が用いられる.可溶化した膜蛋白質は通常の可溶性蛋白質と同様に沈殿剤を使 って結晶化される.こうしてできた膜蛋白質結晶には,二つの特徴がある.1つは結晶格 子形成に不可欠な蛋白質間の相互作用が,界面活性剤に覆われていない親水性領域間の接 触によって行なわれていること,もう一つは,蛋白質と蛋白質の間の空間が,界面活性剤 のミセルによって埋められている点である.我々グループのミヘル教授が,バクチリオロ ドプシン,光合成光反応中心などの結晶の良好な結晶に成功して以来,膜蛋白質の結晶化 は主に界面活性剤のミセルのサイズを調節することによって行われてきた.このために通常,各種のサイズの界面活性剤,及びこれに各種の小さな両親媒性の物質を加えることに よっておこなわれてきた.しかし,いつも膜蛋白質が充分に大きな親水領域を持っていえ とはいえず,ミセルを最適化する方法には限界がある.そこで今回我々は,膜蛋白質の親 水性部分に着目した.図3に我々のコンセプトをまとめた.膜蛋白の親水領域はいま述べ た通りいつも充分に大きいとは限らない.そこで,われわれはこの親水性部分に他の蛋白 質を結合させてこの領域を拡大することを考えた.図の右側の一番極端な例を考えてみると解かりやすい.親水部分が充分に大きくなれば,界面活性剤のミセルのサイズを無視で きる様になり,このような蛋白質からは,可溶性蛋白質同様に結晶が得られることが期待 される.もちろん,あまり大きな複合体を形成すれば,逆に結晶が得にくくなるであろう.これらめ観点から,我々はこの親水性部分の拡大のために,チトクローム酸化酵素に特異 的なモノクローナル抗体のFvフラグメントを作成した.このフラグメントは抗体分子の中で抗原に特異性を持った最小の安定な単位で,VLとVHlの二本のポリペプチドからなって いる(分子量は約28,000).われわれが抗体のFvフラグメントに着目した理由は,1.特 異性が高いこと,2.親和性が高いこと,3.構造が比較的安定なことが挙げられる.わ れわれはパラコッカス由来のチトクローム酸化酵素と抗体Fvフラグメントの複合体を作成し,これより良好な結晶を得ることに成功した.実際の結晶中でのパッキングも,チトク ローム酸化酵素の親水部同士が,抗体のフラグメントを介して相互作用することによって 行なわれていた(文献3を参照されたい).
チトクローム酸化酵素のX線結晶構造解析
本酵素結晶の構造解析の特徴としては,シンクロトロン放射光および巨大分子用ワイセ ンベルグカメラを用いた,高精度のデータ収集とdensity modificationおよび位相拡張の方法にある.以下にはこれらの技術的な側面について解説を加えたい.
本酵素のX線データおよび位相計算について表1にまとめた.本酵素の結晶の空間群は P4で格子定数はa=b=206.7Å,c=83.5Åである.非対称単位に,1個のチトクローム酸化酵 素一抗体Fvフラグメント複合体を含み,溶媒の含有率は76.5%である[3].本酵素結晶は7 種類の異なった重原子を用いた多重同型置換法によって解かれた.重原子誘導体のスクリーニングは研究室の回転体陰極X線発生装置およびR-axisIIを用いて行なった.データは プログラムDENZO[5]を用いて処理し,CCP4に含まれるプログラムのRSPSを使って,主 な重原子の位置を決定した.さらにプログラムMLPHARlEによって実際に位相を計算し, 良好なphasing powerを与えるものだけを選択した.この結果我々は100回程度のスクリー ニングにより, 9個の有効な重原子誘導体を見いだした.実際には最終的に, 6種類の異 なった誘導体から得たデータを位相計算に用いている.表1に示した重原子試薬は塩化水 銀を除く全ての化合物が有機金属である.これらの重原子の結合部位は全て, 界面活性剤 のミセルに覆われた疎水領域のなかに中にあり, ミセルの中を拡散し,結合したものと思 われる.これらの重原子誘導体は非常に良好なパターソン関数のマップを与えた.光反応中心の解析においても同様の傾向がみられており,これらの有機金属試薬は今後の膜蛋白 質構造解析においても非常に有効であると考えられる.また, 実際の反応より拡散が律速 になっていると考えられ, ソーキングの時間も重要な要素になっていると考えられる.
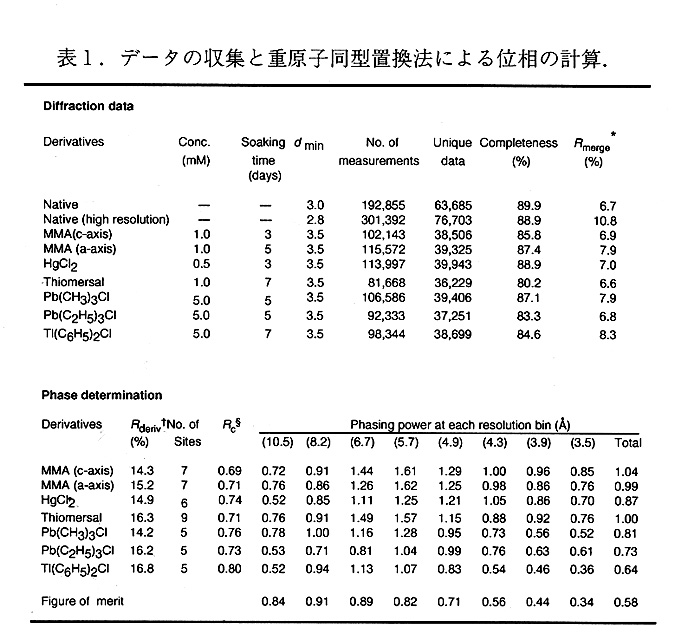
実際の結晶構造解析の計算に用いたデータは文部省高エネルギー物理学研究所の放射光 実験施設(BL6A2)において,坂部知平教授らによって開発されたワイセンベルグカメラ を用いたシステムによって行なわれた.ワイセンベルグカメラを使う理由はいろいろと考えられるが, 特に大きい結晶格子をもつ結晶に対しては, 一度の振動範囲が大きくとれる ことから非常に大きなメリットがある.例えば,チトクローム酸化酵素の場合板に1個の結晶からデータを集めるとすると,ワイセンベルグカメラで3時間程度ですむのに対し通 常の振動写真法では15時間以上かかる.蛋白質の結晶は,測定開始後X線が照射されて いなくても, 徐々に損傷が広がる場合があり, 本結晶は凍結することができないので,振 動写真法では結晶の損傷が激しく良好なデータを得にくい.今回, 多くの重原子誘導体の 結晶を測定する必要もありワイセンベルグカメラの使用が不可欠だった.異常分散効果を利用するために,水銀の誘導体は1.00Å, 鉛の誘導体は0.94Åで測定された.データプロセスには,プログラムDENZOを用いた.位相は異常分散を考慮に入れた多重同型置換法に よって3.5Åまで計算された.このときの平均のFigure of meritは0.58であった.
この3.5Åの電子密度図では, 22本の膜貫通ヘリックスがはっきりと確認され, ヘリック ス間の結合,順序もはっきりしていた.しかし,側鎖は大きなものを除きほとんど確認さ れず,実質的な分解能は5-4.5Å程度と考えられた.側鎖が確認されない理由は高分解能の 反射の位相が, 大きな誤差を含んでいるこ為だが, 10Å付近のアルファヘリックス間パッ キングに由来する強い反射に対し位相がほぼ完全に決まっていて,強くアルファヘリック スの電子密度が出ており,逆にアルファーヘリックス間において電子密度が強く負になっ ていることも原因であると考えられた.実際に分解能5-3.5Åの分解能の電子密度図を描いてみると, 不連続ながら側鎖の電子密度が確認された.そこで,低分解能のデータの重みをへらすことを考えた.単純に,温度因子等を操作して高分解能のデータの重みを増すと,単にノイズの多い電子密度図になるので,有効な中角域だけを持ち上げるような何らかの 関数を考える必要があるが,ここでは,単純にチトクローム酸化酵素の構造因子を一定の 分解能ごとのシェルで,光合成光反応中心のデータにスケーリングするという荒っぽい方 法を考えた.これは,分解能に対する構造因子の平均的な強度分布が,低分解能では強く その2次構造に依存しているという事実による.光反応中心は膜蛋白質であるがチトクロ ーム酸化酵素に比べその膜貫通ヘリックスの含量は半分程度に過ぎない.チトクローム酸化酵素のデータを光反応中心のデータにスケーリングすることにより, 適切にそのヘリッ クスの部分の電子密度を弱めた電子密度を得ることができる.また我々の結晶は強い異方性を示し,aびb軸を含む面では2.8Å以上の分解能のデータを収集可能であるが,c軸方 向では, 3-3.5Åに過ぎない.このままで,電子密度図を描くとc軸方向に強く流れた図しかかけず,非常に解釈が困難であった.これを補正するために,非等方性の温度因子を適 用した.参照になる構造がないため,いろいろな値をあたえて,density modiicationを行なった後,目で電子密度を確認して適切な値を求めた.大変興味深いことには,この適切な値を得たときに,density modificationの結果得たfreeR-factorも最小になった.このことの理論的な背景は明らかではないが,ヒストグラムマッピング法は等方的な原子を前提しているので,それが関係あるのかも知れない.
以上の処理を行なったのち, CCP4のプログラムDMを使って, 3Åまでの位相の拡張を行 なった.density modificationの方法としては,solvent flatteningとhistogram mappingを用いた.最初のfreeR-factorが0.461で200サイクル後には, 0.255まで下がった.この時, 溶媒の含量は75%が最適であった.これによって得られた電子密度図は側鎖等がかなり完全に識別できるものだったが, 電子密度中にしばしば不連続な部分があった.そこで, さらにプログラムSOLOMONを用いて33サイクルのdensity modificationを行なった.この結果得られた電子密度図は連続的で,容易にトレースができるものだった.この電子密度に対して, プログラムOを用いて, モデルのフィッティングを行ないさらにプログラムXPLORを用いて精密化を行なった.現在までに2.8Å分解能までのデータに対して, R因子21.1%のモデ ルを得ている.FreeR-factorは25.6%である.結合距離と角度の理想値からの標準偏差は それぞれ0.01A及び1.8度となっている.精密化された構造はサブユニットIの残基17-554, サブユニットIIの残基1-252, サブユニットIIIの残基1-273, サブユニットIVの残基10-56, VL鎖の残基1-108及びVH鎖の残基1-120の総計1,329残基を含んでいる.
チトクローム酸化酵素の構造
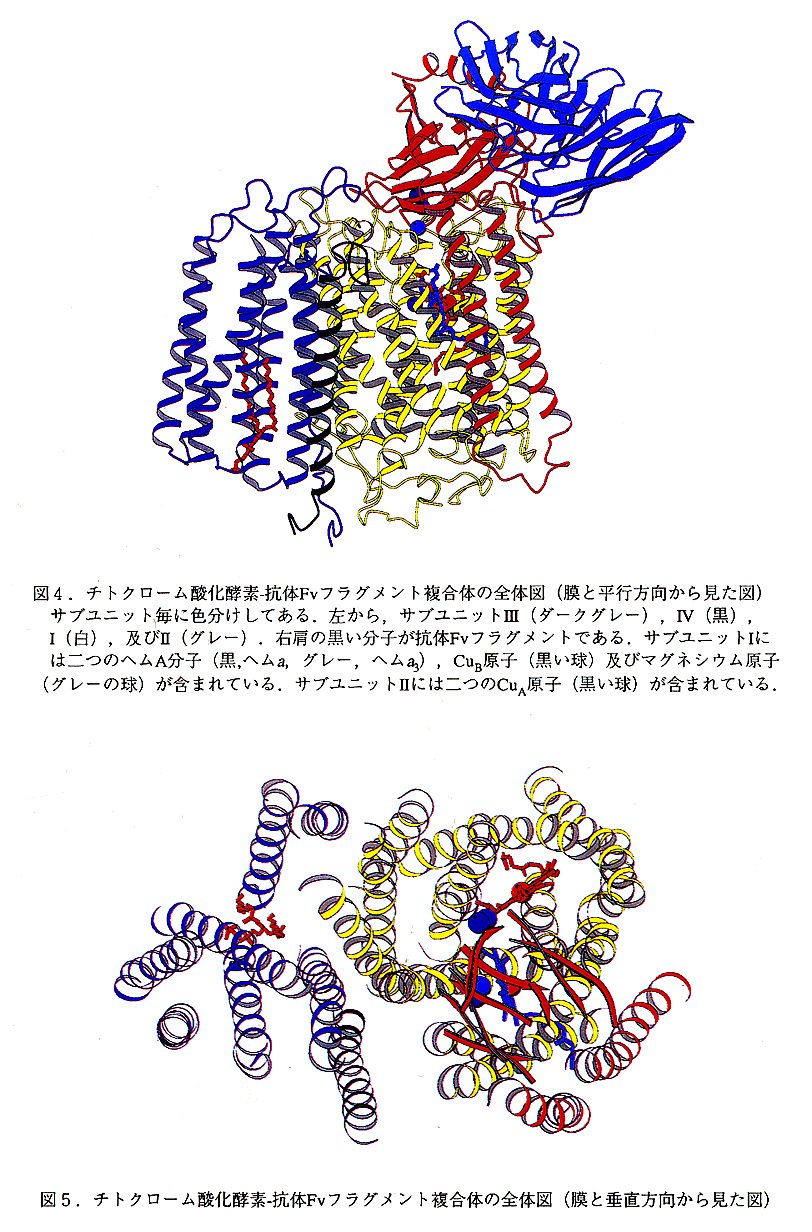
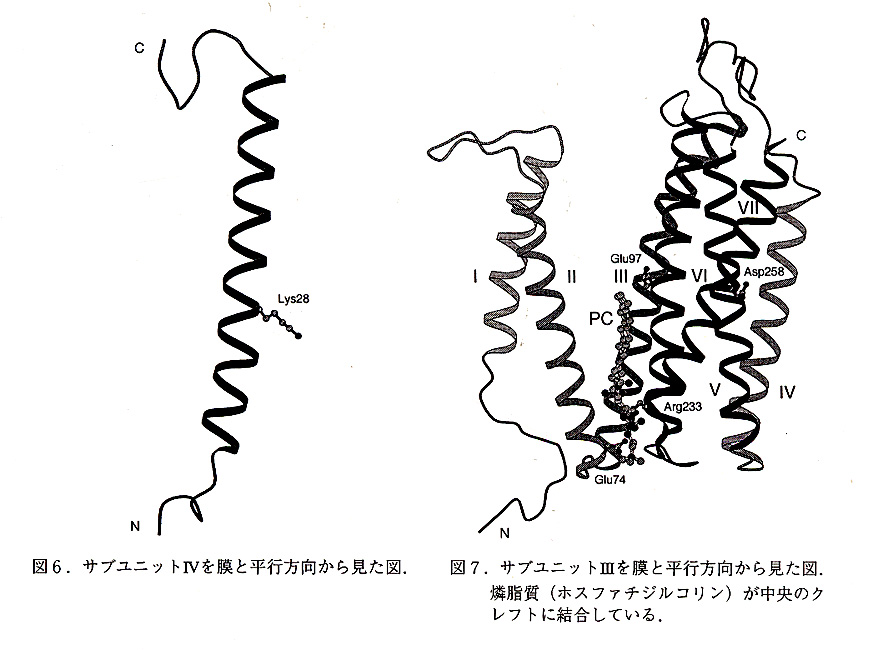
全体構造.パラコッカス由来チトクローム酸化酵素の抗体フラグメントとの複合体の全 体図を図4(膜と並行方向),図5(膜と垂直方向)から示す.膜と並行な方向からみた, 膜貫通部分は台形をしており,そのペリプラズム側は,最大75Å,サイトプラズム側は最 大90Åである.膜貫通部分の高さは最大55Åである.膜貫通部分は22本のヘリックスからなっている.サブユニットIIの球状ドメインが台形の上側についており,この結果,複合 体の膜に体して垂直方向に95Åの長さになっている.複合体の中心部にはサブユニットIがあり,その両側に,サブユニットIIとサブユニットIIIが結合している.サブユニットIIのC末端は,ペリプラズム側で球状のドメインを形成している.抗体のFvフラグメントはこのドメインに結合している.サブユニット1VはサブユニットIとIIIの問に位置している.サブユニットIV.図6はこのサブユニットを膜に並行な方向から見た図である.この 小さなサブユニットの機能は不明である.このサブユニットは56残基からなり一本の膜 貫通ヘリックスを含んでおり,N末端は細胞質側にある.最近になってこのサブユニット の遺伝子がクローニングされ,これが他の蛋白質のリーダーシークェンスであることが解 かった.このサブユニットは,恐らく,この複合体の安定化の働きをしていると考えられ る.サブユニット1Vはただ1本の膜貫通ヘリックスしか有していないが, 興味深いことに 他の三個の全てのサブユニットと接触している.これにより複合体の安定化に寄与している可能性がある.
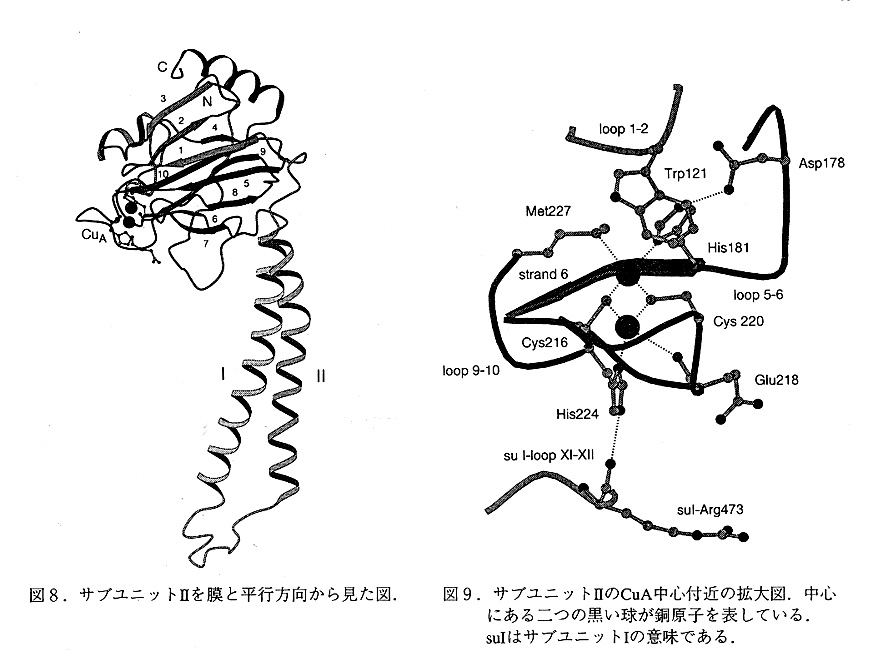
サブユニットIII. 図7はこのサブユニットを膜に並行な方向から見た図である.このサブユニットの機能も不明である.このサブユニットは酸化還元に関わる金属中心も含んでいないし, プロトンポンプの働きにも関係していない.チトクローム酸化酵素はサブユニットIとIIだけで完全に機能することが知られている.このサブユニットはチトクローム酸化酵素の膜におけるアッセンブリに関与しているとも言われている.サブユニットIIIは7本の膜貫通ヘリックスからなっており,二つのドメインに分けることができる.ヘリックスI-IIとヘリックスIII-VIIがそれぞれ別のドメインを形成しており,二つのドメインの間には,大きなV字型のクレフトがある.このクレフトの底にはリン脂質の分子が結合している.現在この分子はホスファチジルコリンと同定されている.このリン脂質のコリンの部分の結合部位Arg233とAsp74によって形成されている.このクレフトは他の膜蛋白質の結合部位である可能性がある.一番可能性が高いものとしては,膜結合のチトクロームc552が挙げられる.パラコッカスにおいて,チトクローム酸化酵素に対する実際の電子供与体はこの膜結合のチトクロームc552と考えられている.
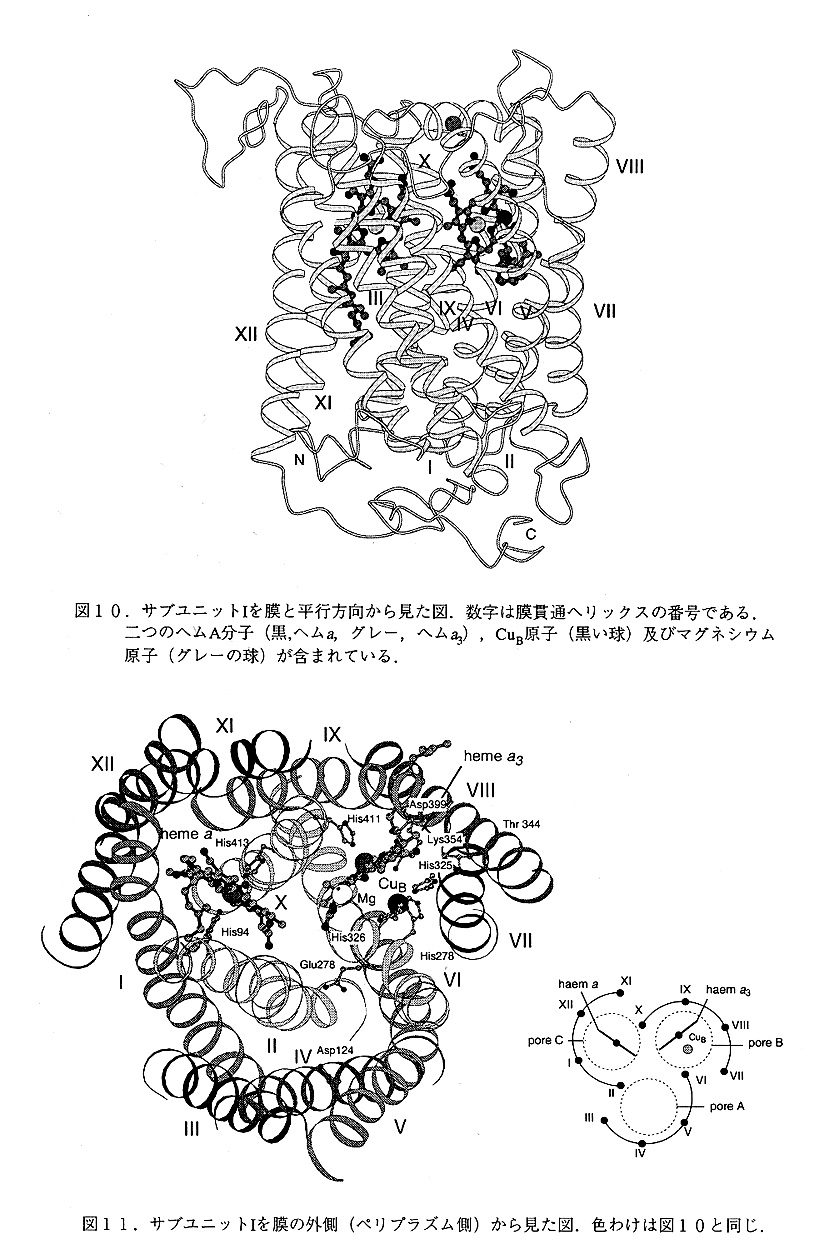
サブユニットII, サブユニットIIは3つのセグメントからなっている(図8).N末端 ループ(残基1-26),二本の膜貫通部位を含むセグメント(残基27-105)及びCuA中心を 含むC末端の球状ドメイン(残基106-252)である.N末端のループとC末端の球状ドメ インは,サブユニットIのポアB(後述)の上にあり, ペリプラズムの側で強く相互作用 している.膜貫通ヘリックスIとIIはサブユニットIの膜貫通ヘリックスIX及びVIIIと強く相互作用している.
C末端の球状ドメインは10本のストランドからなるβバレルで,アズリンやブラスト シアニンなどタイプIの銅蛋白質と同様のホールディングをしている.ブラストシアニン のα炭素の位置をこの構造に重ね合わせると, 99残基の内74残基が2.2Aのr.m.s.dで重ね合わせることができる.C末端の球状ドメインのタイプI銅蛋白質との最大の違いは,ストラ ンド3,ストランド4及びその間のループの部分である.
タイプIの銅蛋白質との最大の違いは,原子価が共に1.5であると考えられる銅原子 (CuA1及びCuA2)を二つ含んだ,CuA中心である(図9).この2つの銅原子の結合部位 は,ストランド6とストランド9と10の間のループにある残基からできている.CuA1に配 位しているのはCys216, Cys220, His181及びMet227の側鎖であり,CuA2に配位しているのは Cys216, Cys220, His181の側鎖およびG1u218の主鎖のカルボニル酸素である.両方の銅原子の配位子は共に歪んだ四面体構造をしている.二つの銅原子は,Cys216とCys220の側鎖 により,橋梁けされておりその距離は, 2.6Åである.銅とイオウの原子は同一平面上にあ り, ちょうど2Fe-2S型のフェレドキシンの様な形をしている.銅原子とシステインのSγ 原子,メチオニンのSδ原子,ヒスチジンのNδ1原子及びグルタミン酸のO原子までの距離は,2, 3, 2.7, 2.1-2.2及び3.0Åである.これらの距離は,タイプIの銅蛋白質の場合とよく似ている.
また,Trp121は, Met227と3.2Åの距離にあり,Asp178はHis188と水素結合をしている. これらの残基はCuA中心を覆う形で溶媒に露出している.これらの残基はチトクロームc からの電子伝達に関与していると考えられる.この領域を含み, サブユニットIIの球状ドメインとおよびサブユニットIのペリプラズム側の平な部分からなる"肩"(図4参照) の部分がチトクロームcの結合部位と考えることができる.これらの領域には, 10個程 度の酸性アミノ酸の残基が存在しており,これらが, チトクロームC表面のリジン残基と 相互作用すると考えられる.
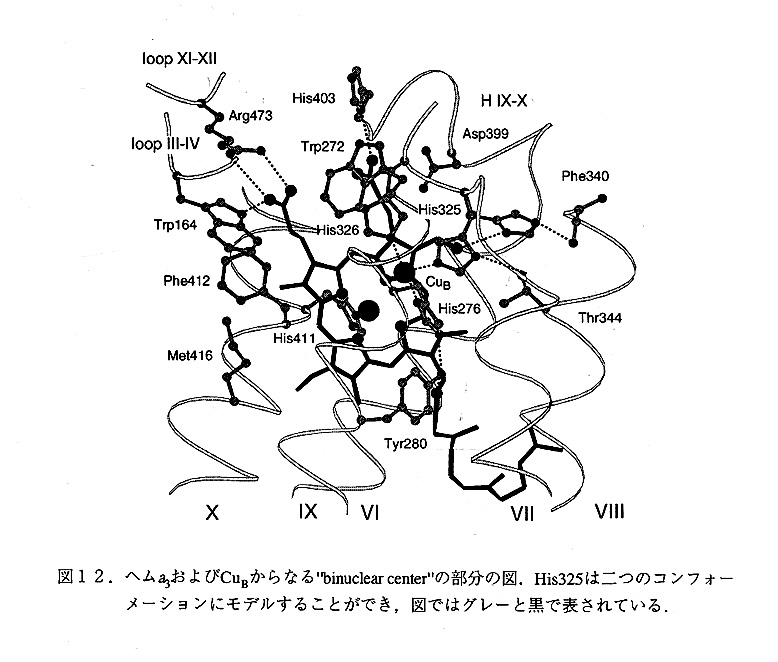
サブユニットI, サブユニットIは全部で12本の膜貫通ヘリックスを持っている(図10). このサブユニットを膜と垂直の方向から見ると,予期しなかったことに疑似3回回転対称 を有していた(図11).ペリプラズムの側からみると,12個のヘリックスが,4本の ヘリックスからなる三個の半円形の弧を形成していて(ヘリックスIII-VI,ヘリックスVII-X,ヘリックスXI,XII,I及びII),ヘリックスII,VI,及びXが分子の三回回転軸に最も近い位 置にあり,ヘリックスIII,VII及びXIが最も離れた位置にある.ヘリックスの並んでいる順番は反時計まわりにシークェンスの順にならんでいる.三個の弧はその一つ前の弧の最後のヘリックスともに,5本のヘリックスからなる"ポア"と呼ばれる構造を作っている. これらのポアはポアA(ヘリックスII-VI),ポアB(ヘリックスVI-X)及びポアC(ヘリッ、ニ クスX-XIIおよびI-II)と呼ばれる。膜を隔てたプロトン濃度勾配を形成するためにはタンパク質のなかに,何の障害もなく,プロトンを通す経路があってはいけない.実際に,三 個のポアは途中でなんらかの方法によってさえぎられており,膜の内側から外側まで直接 通じる経路は見つかっていない.ポアAは途中,よく保存されている芳香環の側鎖によっ てブロックされており,ポアBはheme a3 とCuBをポアCはheme a を有している.12本のヘ リックスは11本のループ(ループI-IIからループXI-XII)で連結されている.ペリプラ ズム側のループの長さは8-26残基であり5-10残基の長さのサイトプラズム側のループより長くなっている.ペリプラズム側のループは内側に折り返すような形になっており,平な 表面を形成している.ループIII-IVとループIX-Xは膜に平行な短いヘリックスのセクションを有している.
N末端とC末端はペリプラズム側にあるが,N末端16残基はディスオーダーして見えて いない.C末端の41残基は,はっきりとした二次構造を形成している部分は少なく,細 胞質側にサブユニットの底を形成するように存在している.
ヘムa, ヘムaは低スピンヘムで,二つのヒスチジン(His94とHis413)がヘム鉄に配位 している.このヘムはCuAからバイニュークレアセンダーへの電子伝達を仲介していると 考えられている.ヘムAは,タンパクに共有結合しておらず,またホルミル基と疎水性の 長いヒドロキシルファーネシル基を持っているのが特徴である.また,ヘムのプロピオン 酸基,ホルミル墓及びヒドロキシルファーネシル基のなかの,酸素原子は全て,イオン結 合または水素結合を形成している.後述するヘムa3とは異なり,ヘムaのヒドロキシルフ ァーネシル基は,ポアCのなかに存在している.ポアCは疎水的な残基で構成されており ヒドロキシルファーネシル基を強く結合している.従って,このポアCは完全にブロック されており,プロトンの経路に使われているとは考えられない.
ヘムa3, ヘムa3とCuBからなるbinuclear centerは酸素を水に還元する反応を実際に触媒 する部分である(図12).これは,ホァBのなかに存在しているが,ヘリックスIXとX の間のループ(実際には膜に水平なヘリックスになっている)も重要な働きをしている と思われる.ヘムa3の軸リガンドはヘリックスXのHis411だけで,もう一方のサイトは開 いている.実際にはこの部分に,溶媒またはアジドと思われる電子密度が存在していて, 5.2AはなれたCuBとの間に橋をかけたような形となっている.現在の電子密度図のなかで は,鉄原子はヘムの平面から,約0.7Aはずれており,ちょうどデオキシヘモグロビンのよ うな形になっている.実際に酸素の結合する位置は溶媒の電子密度の見える位置である と考えられる.His27(Nδ1) と His325 と His326(Nε2)はCuBに配位している. Tyr280は His276と水素結合できる位置にあり,Trp272ははHis326とスタッキングしている.興味深 いのはHis325である.実際の電子密度図の中で,この残基はディスオーダーしており, 二つのコンブォーメーションにモデルすることができる.1つは先述のようにCuBに結合 する位置にあり,Thr344と水素結合を形成している.もう一つはHis325の側鎖が完全に銅 から自由な状態であり,他のカルボニル酸素と水素結合している状態にある.これは母液 中に存在する,アジ化ナトリウムの影響である可能性が強い.最近EXAFSの結果から酸 化状態と還元状態でCuBの配位数が変化することが報告されており,結晶解析の結果との 関係に興味が持たれる.いずれにせよ後述のように,プロトンポンプのメカニズムに関与 している可能性の強い残基であり,阻害剤の有無,酸化還元状態の違いによってこの残基 の構造がどの様に違ってくるのがが興味の持たれる残基である.
プロトンの経路
チトクローム酸化酵素の機能には二種類の異なった, プロトンが必要である.ひとつは, 酸素を還元し水分子を生成するのに使われる"化学的"プロトンでありもう一つは実際に 膜を横切って輸送される, "ポンプされる"プロトンである.ゲニスらによって, Asp124 のAsnへの変異酵素がプロトンの輸送能力は失われるが, 酸素を還元する能力を残してい ることが見いだされて以来, 二つの別々のプロトンの経路があるであろうことが予想され ていた.実際に構造のなかにもこれらの経路に相当すると考えられる, 二つの異なったプ ロトンの経路が見いだされた(図13a).
一つはポアBの中を通る経路である.これまでの変異酵素の実験の結果からは,酸素を 還元するプロトンの経路と推定されている.この経路の入り口は, ペリプラズム側のヘリ ックス1VとVの間のループ,ヘリックスW-V皿の間のループそしてC末端のループがらでき ている親水的な窪みである. ここから,Ser291, Lys354, Thr351, ヘムもの水酸基,そして Tyr280が順にならんでおり, 最終的には,ヘムもの鉄とCuBの間の酸素の結合部位にまで 到達していると考えられる.この中で特徴的な残基はLys354であり,この残基の環境が疎 水的で,脱プロトン化している可能性が強いが, プロトンの輸送時にはプロトン化し何ら かの構造変化によってSer291とThr351の間のプロトンの受渡を行なっていると考えられる. プロトンの経路中に,一種のエネルギー的な障害とも考えられる構造がおかれていること はその機能を考える上でも興味深い.
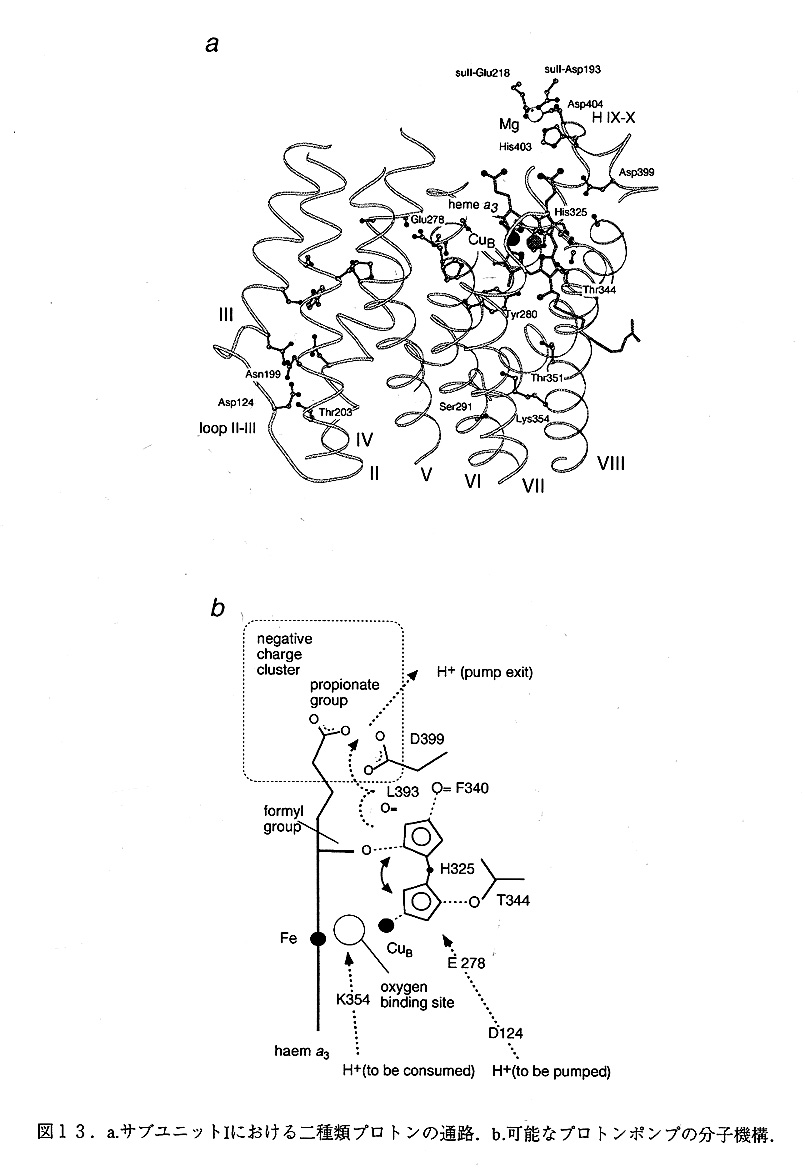
もう1つの経路はポアAの中にある.この経路はヘリックスIIとIIIの間にあるループ近 傍から始まり,Asp124,Thr203及びAsn199が入り口を形成している.ここから,ヘリック スWにあるG1u278まで大きな親水的なチャンネルが延びており,この中にはいくつかの溶 媒分子が確認される.単純な計算ではこの空間に10個程度の溶媒分子をおくことができる. この先の経路は現在のところはっきりしていない.もっとも可能性が強いものとしては CuBの配位子につながる経路だが,これが実現するためには,いくつかの溶媒分子が存在 することが不可欠である. CuBに配位しているHis325まで経路がつながっているとすれば,この先はAsp399から・サブユニットIとIIの間にある親水的なクレフトまで再び大きな親水 性のチャンネルがつながっており,ここが出口となっていると考えられる.この出口の側 に,Asp399,Asp404,ヘムaのプロピオン酸基,サブユニットIIのAsp193,G1u218からなる, 酸性残基のクラスターがありここにMg2+イオンと思われる電子密度が観察される。この 部分の変異酵素の実験から,このMg2+イオンは機能に直接関係がないことが解かってお り,サブユニットIとIIの結合を安定化する働きがあると思われる.
推定されるプロトンポンプの機構
これまで,各種のプロトンポンプの機構が提唱されてきたが実際の構造と照らしあわせ て,現実性を考えてみるとその批判に耐えるモデルは少ない.今回構造解析を行なったた だ1つの状態からは実際のポンプのメカニズムを理解することは難しいが,少なくとも, これまで提唱されているモデルの中から可能1生の高いものを選ぶことはできる.もし,上記に示したCuBを通る経路が正しいとすれば,Wikstromらによって提唱されている,"ヒ スチジンサイクル"のメカニズムが最も現実に近いものであると考えられる.我々は,こ のモデルをべ一スに実際の構造に合うように改変し,さらに則chらによって提唱されてい る,電気的中性保存の原則を取り入れた,"ヒスチジンシャトル"と呼ばれるメカニズム を提唱した.詳細は原典を参照していただきたいが,ここでは,図13bを用いてその概 念を説明したい.この図はbhuc1earcenterの部分を示したものである.二つのプロトンの経路,二つの可能なヒスチジンな位置も同時に示されている.酵素が完全に酸化された状 態で,His325は銅に結合した状態になっていると考えられる.また完全に脱プロトン化し た,イミダゾール酸の状態になっており,この状態はThr344との水素結合により安定化さ れていると考えられる.この状態のbinuc1ear centerに二つの電子がチトクロームから移動してきて,さらに酸素分子が酸素の結合部位に結合する.このとき,電子の負電荷を中和するために二つのプロトンがポンプされるプロトンのチャンネルを通って汲みあげられると考えられる.このときHis325の側鎖は銅から離れ,もう一つの安定な位置に移動して, この時ヒスチジンの側鎖は二つのプロトンを結合したイミダゾリウムの状態になっており,これは他の二つのカルボニル酸素との水素結合により安定化されていると考えられる.この状態で,酸素の還元反応を進行するために,もう二つのプロトンが別の通路から汲み揚げられると考えられる.この結果酸素上の負電化は中和され,1つの水分子を生じる.こ のときbinuc1earcenterの電気的中性を保つため,His325の上に結合していた,二つのプロトンはペリプラズム側に放出され,His325の側鎖はもとの位置にもどる.この様なサイクルを二回繰り返して,酸素分子は完全に水分子に還元され,4個のプロトンが膜を隔てで輸 送されると考えられる.
ここに挙げたモデルはまだ作業仮説の域をでていない.またイミダゾール酸などの化学 的に起こりにくい状態を含んでいる.現在,このモデルを検証および改善するために,酸 化還元状態を変えた酵素などの測定を行なっている。また,現在共同研究者が,プロトン の通路および,CuB中心付近の変異酵素を集中的に作成,解析を行なっている.これらの 結果から・二つのプロトンの通路の役割が確定し,さらに正確なプロトンポンプの機構が 得られることを期待している。
最後になったが,この研究はDeutschMax-P1ank-InstituteにおいてHartmut Miche1教授、 Bemd Ludwig教授(Frankfurt Goethe University)及びChristian Ostemeier博士と共同で行なったものであり,彼等なしには成し得なかったものである。また、放射光実験施設での実験に助言,協力していただいた,筑波大学の坂部知平教授,高エネルギー物理学研究所の渡邊信久助手,鈴木守助手,池水信二研究員に感謝の意を表したい.
文献