1. はじめに
質量分析法(mass spectrometry;MS)は、物質の質量を精確に測定することができ る唯一の分析法であり、現在ではその原理や装置の進歩により、非常に多様な物質 の質量、構造、物理化学的性質を解析する手段として、幅広い分野で利用されてい る。質量分析計は、分析対象物質をイオン化するイオン化部と、その質量を測定す る質量分析部の、2つの部分から構成されている。つまりMSによって測定されるの はイオンの質量である。イオン化された分析対象物質は、質量分析部に於いて電場 や磁場からの作用を受け、その質量1電荷数(masslcharge;m/z)の違いによる運動 能の差に基づいて分離され、質量を測定される。イオン化部、質量分析部のそれぞ れに関して、様々な原理、機構の質量分析計が開発されてきており、得意とする対 象物質も装置の種類によって異なる。
MSは、蛋白質研究の諸領域、特にその質量、一次構造、翻訳後修飾の解析等の分 野に於いても、現在極めて重要な役割を担っている。MSによる蛋白質研究の発展は、 加熱、粒子衝撃、エネルギー照射等の条件を穏和にしたイオン化法の進歩に拠ると ころが大きい。こうした穏和なイオン化法の内、蛋白質の研究に特によく用いられ るのは、エレクトロスプレーイオン化(electrospray ionizatation;ESI)法、マトリック ス支援レーザー脱離イオン化(matrix-assisted laser desorption ionization;MALDI)法の2種類である。特にESI法は最も穏和なイオン化法であり、蛋白質等の生体高分子の 質量を、徴量な試料から高い精度で非破壊的に測定することを可能とした。
ところで蛋白質の高次構造解析法として最も有力な手法は、言うまでもなくX線 結晶構造解析法やNMR法である。一方当然のことではあるが、質量を測定するとい うMSの性質上、これらの方法論のように蛋白質の立体構造を直接的に決定すること は、MSには不可能である。しかしMSと他の分野の技術とを効果的に組み合わせる ことにより、或いは高次構造を保持した状態の蛋白質を直接的にMSで検出すること により、蛋白質のリガンド結合部位を同定したり、動的構造を解明したりすること が可能となってきている。またMSによる蛋白質研究は、必要な試料量が微量で済む という利点もある。このようなことから、蛋白質の高次構造研究分野に於けるMSの 果たす役割もまた重要なものとなりつつある。
MSを蛋白質の高次構造研究に適用する際の方法論は、大きく3つに分けることが できる。その1つ目は、H/D交換とMSとを組み合わせる方法である。蛋白質表面へ の露出度の違いによる重水素化効率の差から、蛋白質のコンフォメーションの違い、 変性過程、表面構造等の解析が可能となる。2つ目は、蛋白質の化学修飾とMSとを 併用した方法論であり、蛋白質の表面構造やリガンド結合部位の解析等に利用され る。3つ目はMS単独で蛋白質の高次構造研究を行う方法である。ESI法は穏和なイ オン化法であるため、実験条件を工夫すれば、高次構造を保持したnative状態の蛋 白質の質量スペクトルを測定することができる。このことを利用して、蛋白質とリ ガンドとの非共有結合性の相互作用や、蛋白質の変性過程を、質量スペクトルから 直接的に解析することが可能となる。本稿ではまず、蛋白質の高次構造研究に最も 適した質量分析法であるESI-MSの原理について簡単に触れる。続いて、上に述べた 3つの方法論のそれぞれについての研究例を幾つか紹介し、MSが蛋白質の高次構造 研究にどのように用いられているのかを概観する。
2. ESI-MSの原理
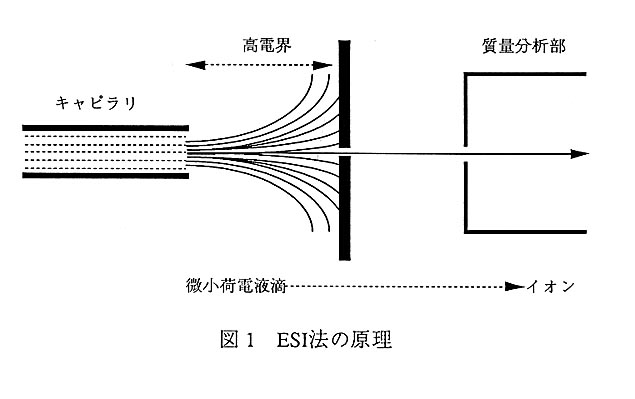
ESI法によるイオンイヒ機構の模式図を図1に示す。ESI法によって蛋白質の質量ス ペクトルを測定する場合には、通常、試料溶液を酸性にして正イオンを検出する。 試料溶液はキャピラリを通して質量分析計内に導入される。正イオンを検出する場 合には、キャピラリの先端出口付近には正電荷の高電界が印加され、そこにおいて 溶液中の正負イオンは空問的に分離される。電荷分離された溶液の内、正電荷に富 んだ部分は、反対電荷の対抗電極からのクーロン引力によってキャピラリの出口か ら引き出され、同軸方向への圧搾窒素ガス流等によって破断霧イヒされる。こうして 生じた微小荷電液滴の表面からは、加熱等により溶媒分子が蒸発し、液滴の半径が 小さくなっていく。最終的に液i滴状態を維持している表面張力による凝集力が液滴 内部の電荷のクーロン斥力に抗しきれなくなると、蛋白質の正イオン(M+nH)n+の自 発的な蒸発が起こり、そのイオンが質量分析部に到達して質量が検出される。ここ で、Mは蛋白質分子の質量、nはその蛋白質のプロトン受容部位の数である。つまり ESI法により検出されるのは、蛋白質の溶液中での荷電状態を反映した多価イオンで ある。また勿論nは特定の条件においてもある程度の分布をもった量であるから、 観測される多価イオンは数一数十種類に及び、各多価イオンは質量スペクトル上で は(M+nH)/nのm/z値で観測される。
質量分析計の、イオン化部に於けるイオン化法の種類、質量分析部に於ける質量測 定法の種類の間には、ある程度の組み合わせの相性がある。イオン化法としてESI 法を採る質量分析計では、四重極型の質量分析部を備えている装置が多い。四重極 型質量分析計は、その電極の形状により、Qフィルター型とイオントラップ型の2種 類に分類される。Qフィルター型では、双曲面の形状をした4本の柱状電極が設置さ れており、それらに数MHz程度の周波数の交流電圧を与えることによって四重極電場 を振動させ、それによってイオンを振動運動させることにより質量測定が行われる。 ESI-MSが蛋白質の高次構造研究に適しているといえるのは、上述したような測定 原理により、対象蛋白質の酵素消化物の分離に用いられる液体クロマトグラフィー( liquid chromatography;LC)と組み合わせた実験系(LC/MS)の構築が容易であり、ま たnative状態の蛋白質を直接的に観測できる等の特徴を有しているためである。
3. H/D交換とMSとを併用した蛋白質の高次構造研究法
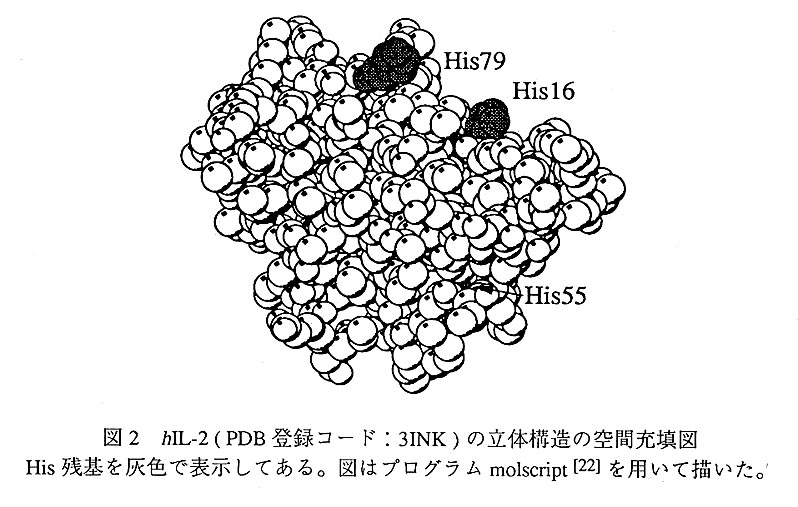
蛋白質を重水(D2O)に溶解しておくと、蛋白質表面に存在している極性の官能基 が重水素化される。蛋白質分子中でHlD交換可能な水素は、アミノ基、カルボキシ ル基、水酸基、ペプチド結合、Argのグアニジノ基に含まれている水素、Trpのイン ドール基の窒素、Hisのイミダゾール基の窒素や8位の炭素に結合した水素である。 表面を重水素化した蛋白質を軽水(H2O)に溶解しなおすと、重水素は軽水素に再置 換されてしまうが、もし蛋白質の表面を重水素ィヒしたまま、質量分析計で測定する ことができれば、蛋白質表面に露出している残基を決定してその機能を解明したり、 蛋白質の変性過程を解析したりすることが可能となる。実験の過程での軽水素への 逆交換をなるべく低く抑えるという観点から、MSとH/D交換を組み合わせた実験に 於いては、適度に交換速度が遅い水素が重水素化の対象となる。ペプチド結合のア ミド水素や、Hisのイミダゾール基のε位の炭素に結合した水素が、それに相当する。 Hisのイミダゾール基の8位の炭素に結合した水素は、中性一塩基性の条件では容 易にHlD交換されるが、酸性条件下では殆ど交換されなくなるので、このことを利 用して重水素化ラベルを保存することが可能である。このHisのH/D交換とMSと を組み合わせた方法が最初に適用されたのは、ヒトインターロイキン-2(hIL-2)中のHis残基の存在環境に関する研究である[1]。hIL-22は免疫応答に関与する蛋白質で、133個のアミノ酸残基からなり、His16、His55、His79の3つのHis残基を有する。 この研究ではまず、hIL-2を塩基性の重水溶液中でH/D交換し、酸性の軽水溶液中でStaphylococcus aureus V8 proteaseにより酵素消化した。次いで、この処理によって得 られたペプチド混合物と、重水素化処理を行わない対照試料の酵素消化物の、高速 原子衝撃(fast atom bombardment;FAB)MS測定を行い、上記の3つのHis残基を含 むペプチド(His16-Asp20、Leu53-Glu60、Val69-Glu95)に関して、両試料の質量 を比較した。その結果、His16、His55、His79各残基のイミダゾール基のε位炭素に 結合した水素の重水素化率は、それぞれ約50%、10%、100%となり、His79は hIL-2の表面に存在し、His55は蛋白質分子内部に埋もれていることが判明した。この後、hIL-2の立体構造がX線結晶構造解析により決定された[2]。その構造を図2に示す。MSによるHis残基の表面露出度の見積りが、ほぼ妥当であることがよく分 かる。
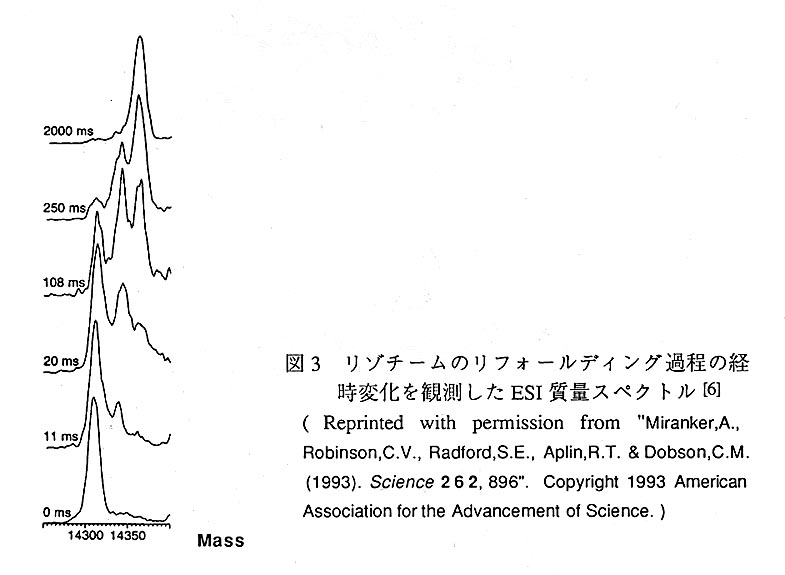
一方、ペプチド結合のアミド水素のH/D交換とMSとを組み合わせた蛋白質の高次 構造研究は、近年盛んに行われるようになってきている。この方法論は、蛋白質の コンフォメーションの違い[3],[4],[5]や、リフォールディング過程[6],[7]の解析に対して特に有効であり、最近ではチトクロームcの表面構造の解析に適用した研究も報告 されている[8]。ここでは、パルスラベル法による/lD交換とESI-MSとを組み合わ せた方法によって、鶏卵白リゾチームのリフォールディング過程を解析した例[6]を 紹介する。この研究では、以下のような方法で実験を進めている。まず、重水溶液 中でリゾチームを完全に変性させ、H/D交換可能な水素を全て重水素に置換する。続 いて、この溶液に10倍容量の弱酸性(pH5.5)の軽水緩衝液(リフォールディングは 起こるがアミド水素のH/D交換は殆ど進まない)を加えて、リフォールディングを 開始させ、経時的にサンプリングを行う。サンプリングした試料に、5倍容量の軽水 緩衝液(pH10.0)を加えて、一定時問の間、蛋白質表面に存在する重水素を軽水素に 置換する。このD/H交換反応を、冷却した0.5M酢酸軽水溶液を加えることにより停 止させ、ESI-MS測定を行う。その結果を図3に示す。リフォールディングを開始し てからの時間τ=0ms のときの質量スペクトルをみると、質量14,313を重心とする 単一のピークが現れている。この時点では蛋白質は完全に変性した状態にあり、ほ ぼ全ての重水素が軽水素に置き換わってしまっていることが分かる。また、τ= 2000msのときの質量スペクトルには、質量14,363を重心とする単一のピークが現れ ている。リゾチームがnative状態になった後にD/H交換されたため、蛋白質の内部 に埋没したアミド水素が重水素のまま残存した分、質量が増加している様子が分か る。リゾチームのリフォールディング過程に於いては、αドメインがまず最初にフォー ルディングした、比較的安定なフォールディング中間体が存在していることが、 NMRの研究で示されている[9]。MSの実験に於いても、τ=11ms~250ms の質量ス ペクトルには、前述した両ピークの中問の質量14,341付近に第3のピークが現れて お;り、一部のアミド水素が保護されたフォールディング中間体が観測されている。 しかもこの中間体の存在比の経時変化もはっきりと現れており、異なったコンフォ メーションを重水素化率の差に由来する質量の差で分離して、その存在比率の定量 まで行えるという、本方法の利点がよく理解できる。
4. 化学修飾とMSとを組み合わせた蛋白質の高次構造研究法
次に、蛋白質の表面を化学修飾し、蛋白質の表面構造やリガンドとの相互作用部位 を、MSで解析する方法について述べる[10],[11],[12]。ある蛋白質とリガンドとの相互 作用部位を本法によって解析する際に、最も一般的に取られる手順は、リガンド存 在、非存在の両条件下で蛋白質の官能基を化学修飾した後、それぞれの酵素消化物 のMSを測定して、ある特定の残基の修飾の有無を解析するというものである。
ここでは、鶏卵白リゾチームとその阻害剤tri-N-acetylglucosamine(NAG3)の相互作 用部位を解析した例を紹介する[10]。この研究で用いられている方法は以下の通りで ある。まず、NAG3存在、非存在のそれぞれの条件下で、リゾチームに選択的な化学 修飾反応を施す。具体的には、Trp残基のインドール基のKoshland試薬による修飾、 N末端及びLys残基のアミノ基のアミジン化、C末端、Asp及びGlu残基のカルボキ シル基のグリシンアミド化の、3種のィヒ学修飾を行っている。反応の進行具合は、 ESI-MSにより分子量の変化を観測して確認する。続いて、各試料中のリゾチームの S-S結合を還元カルボキシメチル化した後、キモトリプシンで酵素消化を行う。最後 に、得られた酵素消化物中の各ペプチドの分子量を、LClMSで決定する。
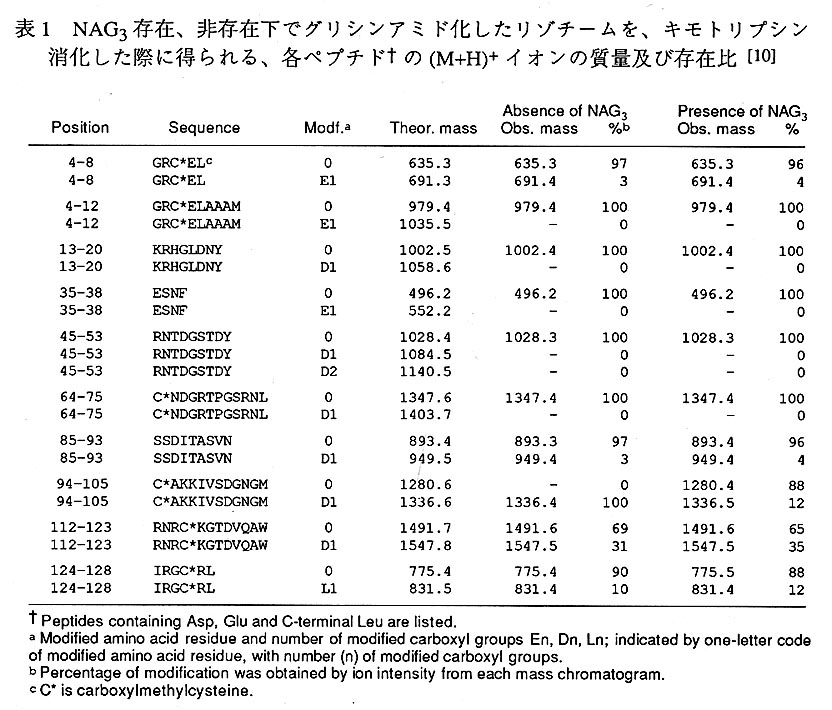
アミノ基のアミジン化に於いては、NAG3の有無による修飾度合いの差は現われず、 何れの場合も、N末端のLys残基を除く殆どのLys残基(Lys13、Lys33、Lys96、 Lys97、Lys116)の8アミノ基が修飾されていた。この結果は、アミノ基はNAG3と の結合には関与していないことを示している。NAG3存在、非存在下で、リゾチーム のカルボキシル基をグリシンアミド化したときの、LClMSの結果を表1に示す。 NAG3非存在下では、Asp101のみが完全に修飾され、Asp119とLeu129(C末端残基) はそれぞれ約30%、10%修飾されているが、他の酸性残基のカルボキシル基は殆ど 或いは全く修飾反応を受けていない。NAG3存在下で修飾反応の受け方に変化があったのは、Asp101のみであり、そのカルボキシル基の修飾率は、100%から12%に著 しく低下している。T1甲残基の修飾反応に於いては、NAG3非存在下では、Trp62のみが修飾され、他のTrp残基(Trp28、Trp63、Trp108、Trp111、Trp123)は修飾さ れなかったのに対して、NAG3存在下では何れのTrp残基も修飾されなかった。これらの結果より、Trp62とAsp101がNAG3との結合に関与している可能性が示唆される。実際この結果は、リゾチームとNAG3の複合体のX線結晶構造解析の結果[13]と一致している。
5. MS単独による蛋白質の高次構造研究法
蛋白質の精確な質量を決定することを目的としてESI-MSによる測定を行う場合、 試科は通常、有機溶媒を含む酸性溶液(例えば1%酢酸、50%メタノール水溶液) に溶解された状態で、質量分析計に導入される。試料を有機溶媒を含む溶液に溶か す理由は、質量分析計内で噴霧させた荷電液滴の表面張力を小さくするすることに より、液滴中の蛋白質イオン間の静電的反発の効果が相対的に強くなり、イオン化 の効率が高められるからである。従って通常の場合は、MSは変性状態の蛋白質の質 量スペクトルを測定していることになる。しかし、ESI法は上述したような穏和なイ オン化法であるため、有機溶媒を含まない溶液に試料を溶解したり、質量分析計の 温度を通常の場合よりも低くする等、実験条件を工夫することによって、native状態、 或いはそれに近い状態の蛋白質の質量スペクトルを測定することも可能である。
このことは、ある蛋白質と、低分子性リガンド、核酸、他の蛋白質等との非共有結 合性の相互作用を、質量スペクトルから直接的に解析することを可能にする[14][15][16]。またESI-MSで蛋白質の質量スペクトルを測定すると、その蛋白質が高 次構造を保ったnative状態にあるのか、或いは変性状態にあるのかという差異によっ て、多価イオンの価数分布が異なってくることが知られている[17],[18]。従ってESI-MS単独による手法でも、蛋白質のコンフォメーション変化や変性過程を解析す ることが、ある程度は可能である[17],[18],[19],[20],[21]。蛋白質のESI-MS測定を正イオンモードで行う場合、変性状態にある蛋白質は価数の大きな正イオンが、native状態の蛋白質はそれと比較してより小さな価数の正イオンが、検出されるのが一般的である。また検知される多価イオンの価数の分布幅は、前者の方が後者よりも広い。 一定の立体構造を保った蛋白質中では、荷電アミノ酸残基が存在する環境に応じた 固有のpKa値をとっていること、また周囲の静電的環境の影響を受けることにより 酸性アミノ酸残基のpKa値が小さくなること等が、こうした現象の原因であるとさ れている。
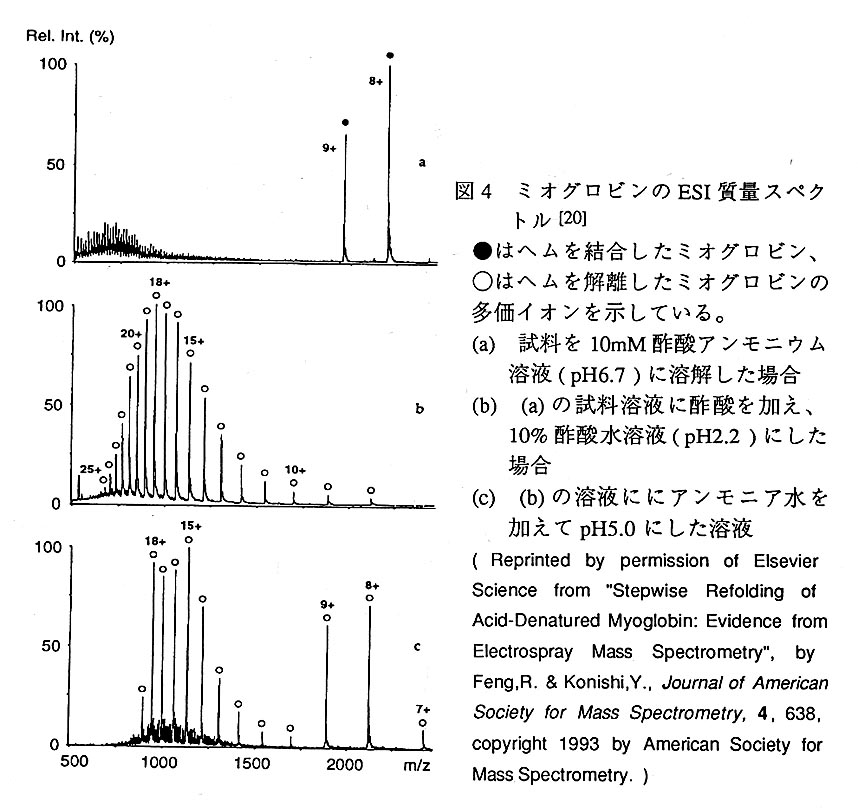
ここでは、上述したような現象を捉えた、この分野に於ける初期の研究例の1つ[20]を紹介する。その内容は馬のミオグロビン(分子量:17568(ヘム有)、16951( ヘム無))の酸変性過程をESI-MSにより解析したものである。図4に同蛋白質の ESI質量スペクトルを示す。ミオグロビンがnative状態を保持し得る条件(pH6.7)に 於ける質量スペクトル(図4a)上では、へムと非共有結合的に会合したミオグロビン の多価イオンが、低価数(+8価、+9価)で検出されており、native或いはそれに近い 状態で同蛋白質が存在していることが分かる。こうした非共有結合性の相互作用の MSによる直接的な検出は、リガンド結合能のアッセイ法等として、蛋白質の生物機 能の解明に対してもESI-MSが有効であることを示している。一方、lミオグロビンが 変性してしまう条件(pH2.2)での質量スペクトル(図4b)上では、へムを解離した ミオグロビンの多価イオンが、高価数かつ非常に広い分布幅(+8価~+25価)で検 出されており、同蛋白質が変性した状態になってしまっていることが分かる。図4c は、ミオグロビンをリフォールディングさせる途上(pH5.0)の質量スペクトルであ るが、変性状態のミオグロビンと推定される高価数の幅広な多価イオン分布と、 native或いはそれに近い状態のミオグロビン(但しへムは保持していない)と推定さ れる低価数の狭幅の多価イオン分布が、bimoda1な状態で検出されている。この結果 は、図4a、図4bの両質量スペクトルに於ける多価イオン分布の差が、PH変化に伴 う蛋白質の荷電状態の変化ではなく、蛋白質のコンフォメーション変化に専ら起因 しているという議論を支持するものである。この研究では、さらにリフォールディ ングを進行させたときの質量スペクトルも示しており、それによると、図4c右側に 現れているような、native或いはそれに近い状態のへム無ミオグロビンの多価イオン が徐々に減り、それに代わって、図4aに現れているような、へムと非共有結合的に 会合したミオグロビンの多価イオンが増加してくる結果が得られている。こうした 結果より当研究の研究者らは、まず蛋白質のみがnative或いはそれに近い状態まで折 り豊まれ、最後にヘムが結合部位に取り込まれる、2段階のリフォールディング過程 をとると結論している。
6. 終わりに
MSによる蛋白質の高次構造研究の概要を以上で述べてきたが、MSと他の分野の 技術と組み合わせることによって、場合によってはMS単独によっても、ある程度の レベルの蛋白質の高次構造研究が可能であることを、ご理解頂けたと思う。確かに 現在のところは、MSによって得られる蛋白質の高次構造情報は、X線結晶構造解析 やNMRと較べると、質、量共にはるかに劣っている。しかし、必要な試料量が少な くて済む利点や、高質量分解能測定が可能なフーリエ変換イオンサイクロトロン共 鳴質量分析計等の装置、技術の進歩に伴い、蛋白質の高次構造研究に於けるMSの重 要性は、今後、益々高まってくるものと考えられる。
参考文献
[1] J Miyano,H., Suzuki,E., Akashi,S., Furuya,M., Tsuji,T., Hirayama,K. & Nagashima,N. (1989). Anal. Sci. 5 , 759.
[2] McKay,D.B. (1 992). Science 257, 412.
[3] Winger,B.E., Light-Wahl,K.J., Rockwood,A.L. & Smith,R.D. (1992). J. Am. Chem. Soc. 114, 5897.
[4] Katta,V. & Chait,B.T. (1993). J. Am. Chem. Soc. 115, 6317.
[5] Gervasoni,P., Staudenmann,W., James,P., Gehrig,P. & Pluckthun,A. (1996). Proc. Natl. Acad. Sci. USA93, 12189.
[6] Miranker,A., Robinson,C.V., Radford,S.E., Aplin,R.T. & Dobson,C.M. (1 993). Science 262, 896.
[7] Heidary,D.K., Gross,L.A., Roy,M. & Jennings,P.A. (1997). Nature Struct. Biol. 4, 725.
[8] Dharmasiri,K. & Smith,D.L. (1996). Anal. Chem. 68, 2340.
[9] Radford,S.E., Dobson,C.M. & Evans,P.A. (1992). Nature 358, 302.
[10] Akashi,S., Niitsu,U., Yuji,R., Ide,H. & Hirayama,K. (1993). Biol. Mass Spectrom. 22, 124.
[11] J Suckau,D., Mak,M. & Przybylski,M. (1992). Proc. Natl. Acad. Sci. USA 8 9, 5630.
[12] Ohguro,H., Palczewski,K., Walsh,K.A. & Johnson,R.S. (1994). Protein Sci. 3 , 2428.
[13] Blake,C.C.F., Johnson,L.N., Mair,G.A., North,A.C.T., Phillips,D.C. & Sarma,V.R. (1967). Proc.R. Soc. B167, 378.
[14] Ganem,B., Li,Y.-T. & Henion,J.D. (1991). J. Am. Chem. Soc. 113, 6294.
[15] Ogorzalek Loo,R.R., Goodlett,D.R., Smith,R.D. & Loo,J.A. (1 993). J. Am. Chem. Soc. 115, 4391 .
[16] Robinson,C.V., Chung,E.W., Kragelund,B.B., Knudsen,J., Aplin,R.T., Poulsen,F.M. & Dobson,C.M. (1996). J. Am. Chem. Soc. 118, 8646.
[17] Loo,J.A., Ogorzalek Loo,R.R., Udseth,H.R., Edmonds,C.G. & Smith,R.D. (1991 ). Rapid Commun. Mass Spectrom. 5 , 101.
[18] Mirza,U.A., Cohen,S.L. & Chait,B.T. (1993). Anal. Chem. 65, 1.
[19] Hutchens,T.W. & Allen,M.H. (1 992). Rapid Commun. Mass Spectrom. 6 , 469.
[20] Feng,R. & Konishi,Y. (1993). J. Am. Soc. Mass Spectrom. 4 , 638.
[21] Hamdan,M. & Curcuruto,O. (1 994). Rapid Commun. Mass Spectrom. 8 , 144.
[22] Kraulis,P.J. (1991 ). J. Appl. Crystallogr. 24, 946.