
構造生物 Vol.5 No.1
¶1999年4月発行
組換え型タンパク質生産方法の現状と問題点
馬場 忠
筑波大学応用生物化学系
1)はじめに
目的の遺伝子を単離・解析すると,次はその遣伝子産物であるタンパク質のより 詳細な機能と構造の解析を目的とした研究計画をたてるのが常となります。また,抗 体作製のためにも組換え型タンパク質を調製することが必要となります。今日のよう に,タンパク質を精製することよりも遺伝子を単離することの方が容易になっており ,そのタンパク質が不安定であったり,組織や細胞にごく微量しか含まれていない場 合などでは組換え型のものを調製するしか方法がない場合が多くあります。私の研究 室では哺乳動物由来の生殖に関係する遺伝子(タンパク質)と植物由来の多糖含成に関 与するものを扱っていますが,大部分のものは大腸菌で発現はしますが生成されたタ ンパク質は不溶性のもので封入体を形成してしまっています。大腸菌の培養温度を操 作したり,不溶性のタンパク質を変性剤で処理したりして,現時点でできる最善の努 力はしていますが,改善が見い出されない場合がほとんどです。そのような時には, 翻訳開始の場所や途中のアミノ酸のコドンを変えたりしないで,別のベクターを使っ たり,大腸菌の系を思い切ってあきらめて酵母や動物細胞の系を使うようにしていま す(それでもしりつぼみになってしまうケースが多いように思います)。ですから,私 は外来遺伝子発現系のプロではありませんし,ひとつの遺伝子を何が何でも活性を保 持した状態で発現させるまで突き詰めて実験をしているわけでもありません。不謹慎 な言い方ですが,「ひとつのベクターを使ってダメならその系はあきらめるや,「数 うちや当たるでいろいろ試してみる方式で実験をしています。しかし,会合で集まら れた方々はうまくいかないから聞きに来た方が多くいるので,「もっと具体例を示し て欲しいとか「どうしてうまくいかないのですか?という要求や質問が出てきます。 これはもっともですが,実は私自身も同じ悩みをもっています。製品のカタログに似 たような遺伝子の発現例が出ていたり,カタログの宣伝文句には何とかなりそうだと いうような感じで書いてありますが,自分の遺伝子がそのようにうまく発現するとい う保証はどこにもありませんし,このように組換え型タンパク質の生産方法について 書けといわれても一般的なことしか書けず,この遺伝子の場合にはこのようにうまく いったとしか書けないことをどうぞお許しください。近い将来,翻訳開始のメチオニ ンの後に塩基性アミノ酸がいくつかあったらうまくいかなかったとか,このコドンが あったらそこで合成がとまってしまったとかいう経験的な積み重ねが特に大腸菌での 発現系では大切なような気がします。
ここでは一般的な組換え型タンパク質の生産方法について復習した後に,大腸菌 での発現系に焦点を当ててその問題点についていろいろ述べてみたいと思います。
2)いろいろな発現系の長所と短所
タンパク質結晶化に必要な条件として,
(ア)じゅうぶんな量の精製タンパク質が得られること
(イ)低コストであること
(ウ)技術的に容易な実験系が組めること
(エ)精製方法が簡便であること
などがあげられます。特別な設備が必要なく,いわゆる通常の実験室で扱える発 現系はやはり大腸菌や酵母を用いたものです。図1には組換え型タンパク質発現のた めの宿主細胞をまとめていますが,当然のことながらいずれの場合にも長所と短所が あります。大腸菌を用いた系は迅速で簡便ですが,例えば真核細胞由来のcDNAを用い ると大腸菌自身のコドン利用頻度と合わないためにタンパク質合成が途中でストップ してしまうことがあります。この発現系のもらとも不都合な点は,生産された組換え 型のタンパク質が正常にホールディングせず「異物として大腸菌細胞内に封入体(イ ンクルージョンボデー)を形成しやすいところです。自慢ではありませんが,哺乳動 物生殖細胞由来の数多くのcDNAを発現させましたが,菌体破砕をした後で可溶性画分 に目的のタンパク質が得られたことは一度もありません。ただし,植物由来の多糖合 成酵素の場合は,分子が比較的大きい(80キロダルトン前後)にもかかわらず可溶性画 分に活性を保持した形で得ることができました。それでも,生産された組換え型タン パク質のおおよそ半分は封入体になっていました。しかし,この封入体を大腸菌から 精製することは容易で,精製した封入体にはほぼ目的のタンパクソしか含まれていま せんので,その変性タンパク質を再生することができれば相当な量のタンパク質を得 ることができます(このことについては後述します)。もうひとつだけ,大腸菌での発 現系で注意しなければならないことがあります。翻訳後の修飾,すなわち組換え型タ ンパク質への糖や脂質の付加などは菌体内では起こらないということです。したがっ て,タンパク質の高次構造や活性の保持などにこのような修飾が必要なものに関して は注意が必要となります。
大腸菌の系でうまくいかない時には,酵母や昆虫細胞,さらには動物培養細胞を 宿主とした発現系が考えられます。いずれの場含も宿主・ベクター系が改良されてき ており,数多くのメーカーから細胞も含めて購入することができます。ただし,ほぼ 上の順番でコストと時間がかかり,さらには特別な実験設備や技術が要求されます。 例えば,動物細胞の場合にはできればそれを扱う特別な実験室が必要ですし,さらに 安全キャビネットや炭酸ガス培養器も備わっていなければなりません。また,細胞の 取扱い方も大腸菌とは比べものにならないほど気を使わなければなりませんし,使用 する培地も血清をはじめとしてかなり高価になります。しかし,目的のcDNAさえうま く発現してくれれば,可溶性の組換え型タンパク質が得られる可能性が高くなります 。翻訳後の修飾が必要なタンパク質も目的にかなっていますが,例えば糖鎖の付加な どの場合,天然型の糖鎖とは違った部位に違った糖鎖がつく可能性があることも考慮 する必要があります。私の研究室でも昆虫細胞の発現系を利用していますが,宿主細 胞の「ご機嫌取りが難しく,購入したメーカーのプロトコールではl週間で発現系が 確立できるとありますが,苦労している割にはうまくいかないことが多くあります( もちろんうまくいっている例も聞いています)。
さらに特殊な発現系として,目的のcDNAを動物や植物個体にトランスジーンとし て組込んでしまい,恒常的に組換え型タンパク質を乳や葉などで生産させてしまうこ とも考えられます。想像される通り,これはかなりおおがかりな研究であり,例えば ,ある種の薬剤としての有用性が高いものについては意義が高くなりますが,通常の ものについては適応できません。
3)大腸菌での発現系の例
ここでは,上で述べた植物由来の多糖合成酵素を例にとって大腸菌での発現系の 実際を説明させていただきます。もちろん,ひとつの例ですので,全体のアウトライ ンが理解していただければ幸いで,各人の工夫などによってさらに簡便で有効な数々 の方法があり得ることはいうまでもありません。
枝つけ酵素(branching enzyme)は,澱粉アミロペクチン中のα-1,6結合を介す る分枝の形成を触媒する酵素です。イネ登熟種子中には,枝つけ酵素アイソザイムが 3種類(RBEl,RBE3,およびRBE4)存在することが明らかになっています。これらの酵 素はいずれもプラスチド移行のためのトランジットペプチドをアミノ末端にもつ前駆 体として合成され,プロセシングされた成熟型RBEl,RBE3およびRBE4はそれぞれ756 ,760,788アミノ酸残基より構成されています。これらのアイソザイムを登熟種子か ら精製することは可能ですが,構造と機能の解析などを行うために大腸菌での発現系 を確立させることを試みました。発現ベクターには,ノバジェン社のpET‐23dを使用 しました。このプラスミドベクターはT7ファージプロモーターをもっており,発現さ せる目的のcDNA断片をこのプロモーターの下流に連結すると,宿主大腸菌(例えば BL21(DE3))により供給されるT7RRBE3をコードする全長c DNAのうちでアミノ末端のプラスチド移行シグナル(トランジットペプチド)を除いた 部分を制限酵素で切断し,平滑末端にした後に,市販のNco Iアダプターを連結させてpET‐23dベクターに組込んだ。NAポリメラーゼにより転写 されるようになっています。このT7RNAポリメラーゼ遺伝子は宿主染色体にあらかじ め組込まれており,Iac UV5プロモーター下流にあるため,培地へのIPTG添加による誘導で発現を制御できる ように考案されています(図2)。pET‐23dの場合には挿入するcDNA断片末端のイニシ エーターメチオチンをコードする制限酵素部位はNco I (CCATGG)ですので,成熟型酵素のアミノ末端の部分を遣伝子レベルで置換してやる 必要があります。RBElの場合は,アミノ末端アミノ酸の隣にNco Iサイトがありましたのでこれを利用しました。RBE3ではアミノ末端近傍で制限酵素 を使い平滑末端にした後に,市販のNco I アダプターを連結させました(図3)。通常よく用いられるPCR法によって,RBE4のア ミノ末端近傍をコードする領域を増幅させながら,5’末端にNco I サイトを付加させて発現ベクターに挿入しました。
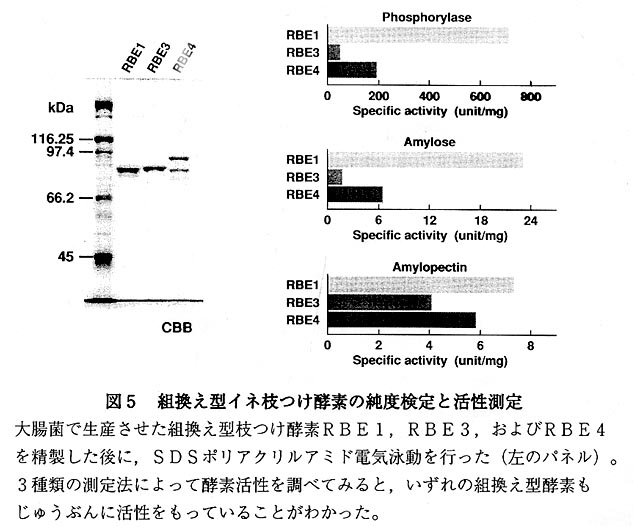
大腸菌BL21(DE3)株をそれぞれのプラスミドで形質転換させて,10}の予備培養を 37℃で一晩行いました。そのうちの8}を新鮮な400}の培地に移して同様に37℃で2時 間培養を行った後に,IPTGを添加して誘導をかけて,30℃で8時間培養を続けました 。菌体を遠心分離器で集菌後,数回適当な緩衝液で洗浄して‐80℃で凍結しました。 可溶性の内容物を得るために,菌体を解凍しながら超音波処理を行い遠心分離で上清 と沈殿に分けました。この上清を出発材料として2回のイオン交換カラムクロマトグ ラフィーを行い,最終的に数10}の精製タンパク質を得ることができました(図4)。も ちろん,いずれの酵素も天然型と同じだけの活性をもっていました(図5)。

4)大腸菌での発現系の問題点と改善策など
前述の通り,大腸菌の発現系で大きな問題となることは,生産された組換え型タ ンパク質が不溶性となり封入体を作りやすいということです。まったく封入体を作ら ないということは,経験的にあり得ないような気もします。これまでに多くの研究者 によって,生産タンパク質の不溶化をできるだけ防いで可溶性画分を多く得る試みや ,不溶性タンパク質を再度活性のある可溶性タンパク質に変える研究が報告されてい ます。
(ア)不溶性タンパク質の生産を最小限に抑えて可溶性のものを得る試みこの目的 のためにいくつかの改善方法が知られています。まず,大腸菌の生育温度を通常の37 ℃から25-30℃に下げて,組換え型タンパク質の生産速度を落としてみることです。 小スケールの実験系で,培養温度を下げ時間をふってやり,可溶性タンパク質の生産 量を調べてみてください。また,発現誘導(例えばIPTGなど)の濃度を下げたり,37℃ で培養するにしても誘導時間を短くしたりするのも効果的な時があるようです。培地 のpHを上げて効果的であったという例もあります。
(イ)不溶性タンパク質の再生
一度生産されてしまった不溶性のタンパク質を再生する方法もいろいろと考案さ れています。精製した封人体を8モル尿素や6モル塩酸グアニジンなどの変性剤で可溶 化し,段階的にそれらの変性剤を透析で除いていく方法によって,タンパク質をリホ ールディングさせた例があります。私達も不溶性になってしまったイネ枝つけ酵素を 変性剤を使って再生しようとしましたが,その酵素自身,変性剤処理しただけで不活 性になってしまうために無駄な試みでした。また,変性剤で安定なタンパク質も可溶 化させることができましたが,活性までは回復しませんでした。そのほかの不溶性タ ンパク質の再生法として,シャペロニンであるGroEで変性タンパク質の立体構造形成 を補助して活性を回復できた例もあります(タカラ社などより購入可能)。また,タン パクジスルフィドイソメラーゼ(PDI)やチオレドキシン,ピューロチオニンなどで, 変性タンパク質のジスルフィド結合を正常なものに戻してやることも考案されていま す(タカラ社などより購入可能)。
(ウ)プロテアーゼ欠損宿主細胞の利用
発現プラスミドに問題がないのにもかかわらず,組換え型タンパク質がまったく 生産されない場合があります。その理由のひとつには使用コドンや使ったDNAの配列 上の間題点(高次構造など)がありますが,生産されるタンパク質が非常に不安定な場 合などでは,大腸菌自身の細胞内プロテアーゼによって分解されてしまった可能性も あります。このような時にはプロテアーゼであるIonやompTの欠損株や変異株を宿主 細胞として使用すると,目的のタンパク質生産がかなり改善される場合があるようで す。
(工)融合タンパク質としての発現
目的のcDNAがうまく発現しない時には,マルトース結合タンパク質やグルタチオ ンSトランスフェラーゼなどとの融合タンパク質として発現させてみるのもひとつの 手段です。通常は,目的のcDNA断片がC末端側になるようにするのが普通です。この 場合,ふたつのタンパク質の間にはエンテロキナーゼやトロンビン,ファクター工な どのプロテアーゼ認識配列ができるように工夫されていますから,精製タンパク質を これらのプロテアーゼで切断すれば目的のタンパク質が単離できるようになっていま す。しかし,この融合タンパク質の系もものによっては封入体を作ってしまいますし ,プロテアーゼが切断しにくい(効率が悪い)場含もありますから,それらのことを念 頭に入れる必要があります。うっかりすると,そのプロテアーゼの購入代だけでもの すごい額になる危険性もあります。
5)おわりに
発現させるcDNAによっては,ほんの一部分ですが可溶性のタンパク質を生産する 場合があります。その時には上で述べた試行錯誤を行うよりも,大量培養をくり返し て精製する方がかえって早い時もあります。私が存じ上げている先生も10リットルの 培養系でわずか一回の活性測定ができる量しか組換え型タンパク質が得られないにも かかわらず,それをくり返すことによって満足のいくデータを作られた福烽「らっし やいます。人の顔がすべて異なるように,用いるcDNAの配列もほとんどの場合異なっ ているのが常です。その場,その場の試行錯誤での判断力の有無と,「石にかじりつ いてもという強い気持ちが成功を産む秘訣なのかもしれません。皆さんの成功を祈り ます。
大腸菌での発現系を中心として思い当たるままを書いてきましたが,ビギナーか らそれ専門のプロの方の間くらいを対象としました。プロの方には「当たり前のこと を書くなと怒られそうですが,どうぞお許しください。また,あえて参考文献を載せ ませんが,近くの書店にいけばこのたぐいの詳しく優れた実験書が数多くありますの で参考にしてください。ここでは,羊土社実験医学別冊バイオマニュアルシリーズ4 巻「遺伝子導入と発現・解析法(横田崇,新井賢一編集),7巻「分子生物学研究のた めのタンパク実験法(竹縄忠臣,稲垣昌樹編集),および10巻「酵母による遺伝子実験 法(山本正幸編集)を参考としています。
第3回坂部プロジェクトジョイントセミナー(1998年7月7日)の講演内容の一部 。他の講演内容は「構造生物」vol.4 No.35-39(1998)に掲載されている。