はじめに
X線構造解析において最も時間と労力が費やされるであろう位相付けの段階で、重原子同型置換体の1つとして最近注目を集めている不活性気体(希ガス)の利用について、その歴史的背景を概観した後、筆者の研究室で実際に行っている実験手順や注意点などを述べ、最後に今後の課題等をまとめてみたい。
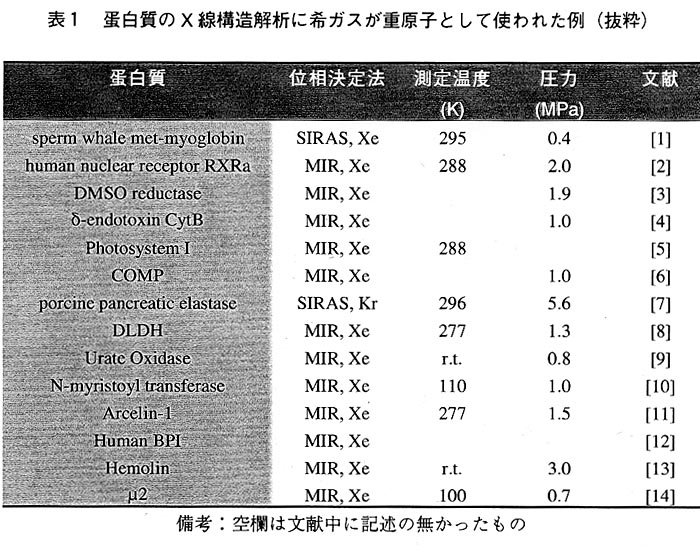
最近になって注目を集めだした背景としてまず第一に挙げられるのは、Kr,Xeのような希ガスを用いて単結晶に物理的な圧力をかけることにより同型置換体を作成し、そのような誘導体を使って実際にX線構造解析に成功した例が数年:前から文献中に頻繁に見受けられるようになったことである(例えば表1を参照)。1995年以前はほんの数例しかなかった報告が、1996年から急速に増えている。これは、Kr,Xeを高圧ガスの状態で簡便に蛋白質結晶に導入する装置の改良が進んだことはもとより、(ある一定の範囲内で)波長を自由に選択できるシンクロトロン放射光や、蛋白質結晶の放射線損傷を最小限に防ぐ極低温下での回折実験が一般的に広まったことと無縁ではないだろう。日本国内で重原子同型置換体作成の一手段として、ルーチン的に希ガスを用いている研究グループは少数であると思われる。しかし、ソーキング法や透析法のような重原子試薬を液体に溶かして同型置換体結晶を得るこれまでの実験と比べ、簡便、かつ短時間で調製可能な希ガスの利用は、今後、あらゆる蛋白質の構造解析で試してみるべき一般的な方法になるであろう。
歴史的背景
希ガスの中でKr,Xeは蛋白質X線結晶学に用いられることの多い原子で、電子数はそれぞれ36,54である。文献として最初に登場した時期は非常に古く、1946年のことである。当時、2つのグループがXeは麻酔効果を持つと主張し(15,16)、1951年、Cullen,GroseのグループがXeは実際にヒトで麻酔効果を持つことを確認した(17)。このような経緯から希ガス(特にXe)は、蛋白質の構造解析における重原子誘導体としてよりも、血液中での溶解度といった性質を詳細に調べるために使われたのが始まりである。それらの実験報告からXeはへモグロビンやミオグロビンに可逆的に結合することが示されたため、Schoenborn,Kendrewらのグループは、実際にへモグロビンやミオグロビンのどの部位に結合するのかをX線構造に基づいて調べ1965年に発表した(18,19)。この時、初めてXeが蛋白質分子内に部位特異的に結合する事が示された。このSchoenbornらの文献は、差フーリエ図からXeの位置を同定し、どのようなモードで結合しているかを明らかにするにとどまり(もちろん、この結果は十分に画期的であった)、Xeを重原子誘導体として位相付けに利用することは想定されていなかった。また、Xeをどのような装置を使って結晶内に取り込ませたのかに関する詳細な記述は見当たらないが、測定はXeガスを数気圧(0.l-0.2MPa)の圧力をかけて充満させることのできる特別なカセットを用い、カセット内のXeによるX線の多大な吸収との戦いであったことが記されている。
希ガスXeを重原子誘導体として蛋白質の位相付けに使える可能性を示したのはSchoenbornとFeatherstoneで1967年のことである(20)。その後しばらくの間、目立った論文発表はなかったが、1984年Tnton,Petskoらのグループが詳細に蛋白質内部の空隙を調べるプローブとしてXeを使い1.9Åという高分解能で構造を議論した(21)。Xeの異常分散効果を考慮した単一同型重原子置換(SlRAS)法による位相付けを初めて試みたのはTntonらのグループで(1)、差パターソン図のハーカー面にはXe-Xeの明瞭な自己ベクトルが示されている。1994年には同型Xe誘導体の調製に関する文献が発表され(22)、最大5.0MPaまで圧力をかけることのできる装置を用い、常温下でクォーツキャピラリー(厚さ0.01-0.03mm)を用いて圧力(0.3-0.5MPa)をかけながら測定を行い、ミオグロビン、ヘモグロビン以外の蛋白質のX線構造解析に成功した。その後、表1に示すように多様な蛋白質のX線構造解析に重原子同型置換体として用いられるようになった。
Xe、Krの性質・物理的特性
X線構造解析で参考になると思われるKr,Xeの諸性質を表2に、線吸収係数を図1にまとめた。Kr,Xeの溶解度を見ると、XeはKrより格段に溶けやすいことが明らかである。またXeのCuKα由来(1.5418Å)の線吸収係数が非常に大きな値を取ることから、実験室系の装置で測定を行う場合、常にこのことを念頭に置いておく必要があるだろう。
Kr, Xeの蛋白質との相互作用は主にVan der Waals力によるので、一般的な重原子試薬による同型置換体の場合と比べ、その結合位置は異なることが多い。典型的な例として、Kr,Xeは粒状蛋白質内部に存在する疎水的空隙に結合することが挙げられる。また、蛋白質活性中心のリガンド結合部位に、ほとんど構造変化なしに結合することも分かっている(27)。結晶格子に依存して、分子間相互作用位置に結合することもある。


サンプル調製およびデータ収集の実際
一般的に希ガスは重原子試薬と異なり安定で扱いやすく、結晶化に用いる緩衝液の組成に依存しない。しかも水溶液中で高い溶解度を示す(273Kで241cm3、293Kで108cm3。いずれも0.1 MPaでの値)。従って、ルーチンワークで迅速にサンプル調製ができる。ここでは筆者の研究室で行っている希ガス(Xe)同型置換体の作成手順を簡単に述べる。
測定は極低温下(100K)で行うので、抗凍結剤の最適化を行っておくことが望ましい。Xeの溶解度を上げるためにSauerら(24)は269Kで圧力をかけることを推奨しているが、筆者の研究室では耐圧器(図2参照)を事前に冷やしておくことで対処している。使用している耐圧器は4Dxray Systems製(25)で大きさは直径約6cm、高さ約8cmと非常にコンパクトなものであるが、他にも市販品があることを付け加えておく。
以下の手順は暫定的なものであり、現在のところ試行錯誤で最適な方法を考えている。
①Xeガスボンベ、耐圧器、顕微鏡をX線発生装置の周りにセットアップする。
②耐圧器の中央部分にバイアル(NALGENECryogenicVialsl.2mL)をセットし、その中に500-700甘Lの結晶化溶液を入れておく。この操作は加圧時に結晶が乾燥してしまうのを防ぐために必須である。
③抗凍結剤入りの単結晶をcryo一loopにすくい取り、耐圧器の中心に置く。
④耐圧器の蓋をして側面のネジ(脱圧力用)がきちんと閉じているか確認する。
⑤0.8-1.0MPa(8-10気圧)で20分加圧後、耐圧器側面のネジをゆっくり回し圧力を抜く。この時、内部の溶液が飛散しないように注意する。
⑥ガスを抜いて15-20秒後、単結晶を凍らせて測定を始める。
上記手順の⑤は最初に試みる場合の基準としての数値で、定常的に単結晶を準備できるなら圧力をかける時間を変えて測定を試みる方がよい。いくつかの系での実験報告もある(例えば文献22,24)。手順⑥の脱圧力後に結晶を凍らせるまでの時間について、結晶中の溶媒からXeが抜けていくと同時に、蛋白質結合部位からも抜けていく(結果として占有率が下がる)であろうことは十分に推察できるので、できるだけ早く凍らせた方が良いと考えられる。しかし、系統的な実験報告が少ないため、早ければ早い程良いとは一概に言えない。脱圧力後、凍らせていない条件下で30分後でもXeが結晶内の特定部位に存在した例もある。このような例は極端かも知れないが、脱圧力後15秒から20秒程度では、少し占有率が下がるかも知れないという程度で測定が行えることが示されている(24)。高圧における水和の問題に関して、現在実験を行っている1.0MPa程度の圧力と分単位の時問では、それほど深刻な問題になっていないように思えるので、今のところ特に考慮はしていない。Xeの水和に関する実験は古くから行われているが、最近の文献でも見ることができる(22)。
現在までに、筆者の研究室では重原子誘導体の1つとして希ガスを用いた新規蛋白質の構造解析に成功していないが、研究室内ですでに構造解析に成功した蛋白質を用いて系統的な実験を行い、これまでの重原子同型置換体の検索と比べてみると、「Xe導入に際し、その調製は結晶化溶液に依存しない」、「ほとんどの場合native結晶と同型を保っており格子定数の変化は見られない」などの利点を実感することができる。
希ガスを用いたX線構造解析の今後の展望
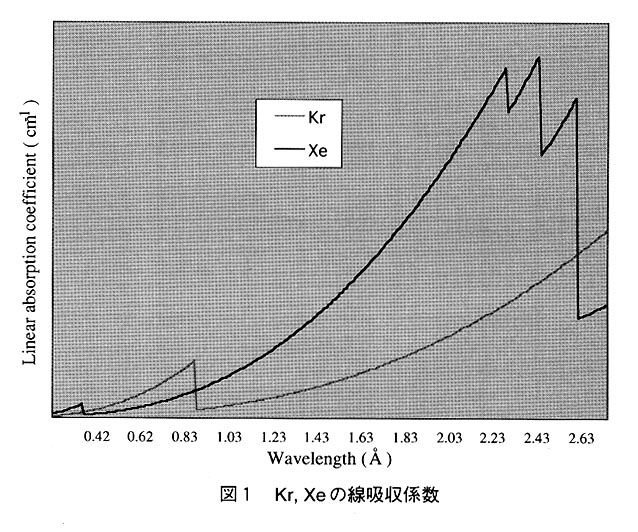
希ガスを重原子同型置換体として扱ったX線構造解析の文献を調べると、圧倒的にXeが多く使われている。これはKr,Xeの電子数やVan der Waals半径、さらには溶解度の差によると考えられる。ある圧力下で同じ数の原子が特定の位置に付くならば、当然、電子数を多く持つ(すなわち散乱能の高い)Xeの方がKrよりも位相計算で多くの寄与をするからである。また、KrはXeより約10倍近くも高い圧力をかけなければならない。つい最近までキャピラリーで圧力をかけながら測定を行っていたことを考えれば、これは非常に不利な点となる。
しかし、KrのK吸収端近傍の波長(0.86552Å)は放射光施設で一般的に使われることの多い波長領域であり、異常分散データを効果的に集めることができる。Krは重原子置換体としてよく用いられるSe(原子番号34、原子量78.96)と元素周期表で同一周期にあり異常分散強度も類似している(図3参照)。従って、データ測定およびその後の処理はSe置換体と同様の取り扱いが期待される。すなわち、安定に稼働している第2世代放射光施設を使って、Se置換体のように多波長異常分散(MAD)法などが適用できる。実験室系でもCuKα由来のX線波長(1.5418Å)における吸収も少ないことから、今後、Krの利用は一層加速すると思われる。
一方、Xeは前述したようにCuKα由来のX線波長ではKrと比べて大きな線吸収係数を示すが(図1参照)、f"=7.348の値から異常分散データを効果的に集めることができる。多大なX線吸収は極低温下の測定、すなわちキャピラリーではなくナイロンループ等を使うことで回避可能である。
第3世代の放射光施設を利用すれば、XeのK吸収端(0.35840Å)より短波長側での測定も可能になり、実際にその可能性が示唆されている(26)。この波長領域では既存のイメージングプレート(IP)やCCDのようなX線検出器の有効性を吟味しなければならない。Schiltzら(26)はESRF BL2/ID11の測定で、ESRF検出器グループが開発したCCD+XRIIを使っている。彼らは様々なIPシステムでもチェソクを行い、CCD+XRIIと同様に使えることを報告している。国内でも第3世代放射光施設Sprhng-8が稼働しており、XeのK吸収端を利用した測定も実験段階であるが報告されている。
まとめ
希ガスは多重同型置換(MlR)法における一同型置換体としてPhotosystem I のような超分子複合体に用いられている一方で、異常分散を考慮した単一同型置換(SlRAS)法にも使われている。非対称単位中にXe 1 原子でも、分子量約85,000のDMSO-reductaseの構造解析に寄与があることから(3)、十分に占有率が高くなれば、分子量約90,∞0の蛋白質にも適用できると見積もられている(28)。Krの場合、非対称単位中に1原子で占有率が半分でも、SlRAS法で分子量約26,000のporcine pancreatic elastaseの構造解析に成功している(7)。高エネルギー領域(XeのK吸収端近傍)での多波長異常分散(MAD)法の利用も現実的になりつつあることから、今後、希ガスを用いて構造解析を成功させる例を、より頻繁に目にすることになるだろう。
参考文献