川上 善之
エーザイ株式会社筑波探索研究所
はじめに
今でもCADD(Computer-Aided Drug Design)というと、後付けの学問と思っている研究 者も多くこの分野に籍を置いている筆者としては閉口している。本物のメディシナルケミストはそのようには思っておらず、いかに上手くCADDを活用して望みのドラッグデザイン(医薬分子設計)を行うかを考えてくれている。ドラッグデザインとは、本来、薬効を発揮する化学構造の仮説を考えて、実際に合成した化合物で証明するという過程の繰り返しであり、その際理論的な根拠を持つことが重要である。なかでもラショナルドラッグデザインは可能な限り少ない化合物の合成で薬物設計を完成させる試みであり、今までに多くの成功例がある(1)。
かつてはバクテリアや実験動物の全身を使ってスクリーニング(薬効評価)をしていたので、そのまま、または少しの構造変換で薬となる物質を天然から、または偶然に発見していた。さらに、先行品の欠点をなくする構造変換で改良型医薬品を発明するいわゆる「ミーツー」研究を行うことが多くあった。
しかし、生体メカニズムに基づいて創薬を行うようになった現在では、HTS(High-Throughput Screening)を利用して入手しうる、あるいは合成したあらゆる化合物(化合物ライブラリー)について、種々なスクリーニングを行うようになった。ただこの種のスクリー二ングは、当然、失敗も多く費用も莫大である。幸運にもこの方法で生物活性物質が見い出されれば、さらにこれを手がかり物質として化学構造を修飾し、構造変換などを行ってリード化合物に高めることを行う。
ターゲットがタンパクや核酸などの生体高分子で、低分子化合物でタンパクータンパク相互作用などを制御する場合には真似るべき低分子化合物がない。低分子リガンドまたは基質がある場合でも、高い選択性が要求され難易度が高くなっている。このように、リード化合物の創出が難しいためターゲット分子の三次元構造に基づく理論的な薬物設計の重要性が増してきている。
構造生物学とSBDDとCADD
構造生物学とは、X線結晶構造解析、NMR、電子顕微鏡などの物理分析手法を用いて、遺伝情報を基につくられ生体細胞内で重要な反応をつかさどっているタンパク質分子の三次元構造を原子レベルで決定し、その立体構造と機能との関係を分子レベルで理解することによって、生体内反応の分子機構を解明することである。さらに、MO,MM,MDなどの計算化学手法を用いて静的および動的構造を解明し体系化するものである。技術でいえば、遺伝子工学・蛋白科学・構造科学・計算化学の共同作業といえる。
1980年代後半からDNAやタンパク質などの立体構造が続々と決定されるようになった。1次構造だけでは解けない、生体高分子の機能の解析が可能になり、データベースの拡充によってアミノ酸配列からのタンパク質立体構造の予測も可能になるなど生物学は新しい時代を迎えようとしている。博物学の時代におこなわれた多くの生物種の記載と同様に、分子"種"の記載によって,フォールドといった新たな一般的原理がうまれてくるであろう。
それは"鍵"としての薬物分子が作用する"鍵穴"(生体高分子)の立体構造の解析技術が進んでいるということである。そこで標的タンパクの構造データをもとに"鍵穴"の立体構造を知り,その形にあった薬物分子をデザインするのがStructure-Based Drug Design(SBDD)である。
"鍵穴"というのは、単にタンパクの結合領域の三次元構造をいう訳ではなく、その疎水性・親水性といった物性や化学的反応性、電気的性質、動的な構造など薬物分子設計に役立つ構造情報全体からなるものである。CADDは標的タンパクの三次元構造の特長を"鍵穴"という構造情報に変換する手段である。つまり構造とSBDDを結び付ける技術である。 この"鍵穴"を参考にメディシナルケミストを中心に薬物設計を進めていき、設計・シミュレーション・合成・構造評価また設計というサイクルをまわして良いリード化合物を見出すことがSBDDの目標である。
CADD技術は、薬物設計者に標的分子の薬物結合部位での薬物との相互作用を明視化し、それらの相互作用を詳細に分析してそれを形成する力を評価する手段となる。視覚化と分析により一連の化合物群における結合の違いや、結合の結果生じた標的分子のコンフォメーション変化を知ることが出来る。
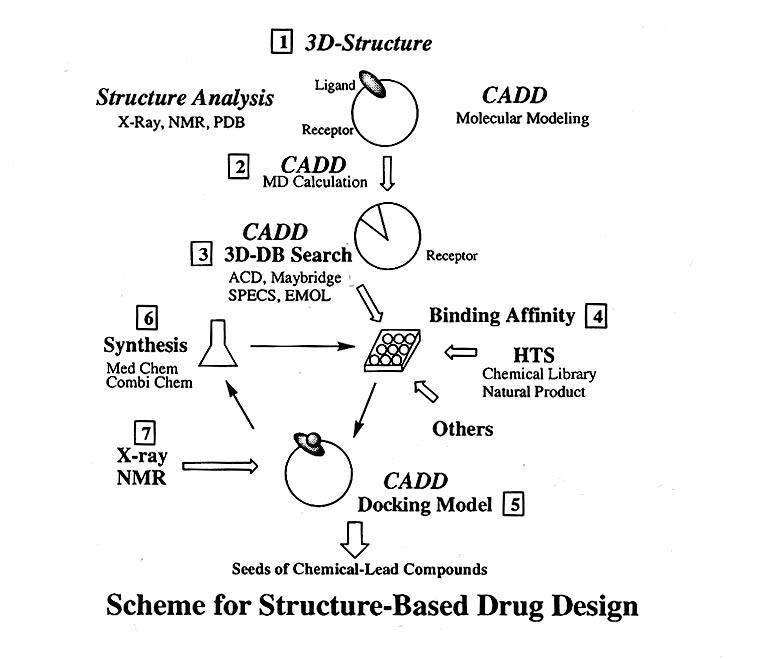
以下に我々が行っているSBDDのスキームを説明する(図参照)。
SBDDの手順
三次元構造…X線結晶構造解析またはNMR解析といった実験により得られた構造(PDB登録構造を含む)、または分子モデリングにより計算から求めた予測構造を初期構造とする。現在までは、小さなドメインを除き構造のほとんどがX線結晶構造解析により得られている。構造解析できない膜存在型タンパクや、配列相同性が高いファミリータンパクについてはホモロジーモデリング法などの分子モデリングにより予想構造を作製する例が増えてきている。
分子動力学計算…1で得られた構造はそのまま使わず、MD計算により動的な構造
や予想結合部位の鍵穴情報を分析する。
シード探索一三次元データベース探索(コンピュータスクリーニング)、HTS、対照薬ほかなどで活性化合物を見つける。
活性測定一薬物と標的タンパク間の結合能の増減を見る。
ドッキング一薬物の標的タンパクヘの結合状態を予想し、構造変換のための更なる
結合部位を探索する。
薬物設計…ドッキング図を参考にして構造を展開する。
複合体構造解析一実際の結合状態を観測して構造を評価することでドッキングシミュレーションに役立てる。
現在我々のところでは複数のテーマでSBDDを行っているが、X線結晶解析やNMRといった構造解析が追いついておらず分子モデリングが先行してドラッグデザインに使われている。しかし、分子モデリングによるデザイン仮説には構造解析実験による実証が不可欠である。
我々が行ったアセチルコリンエステラーゼ阻害剤の研究を例にとると、阻害剤と酵素の複合体のX線結晶構造解析がなされた系を使って、よく構造活性相関が分かっている酵素阻害剤の検証においても分子モデリングの不完全さが認められた。この事を少し示しておきたい。
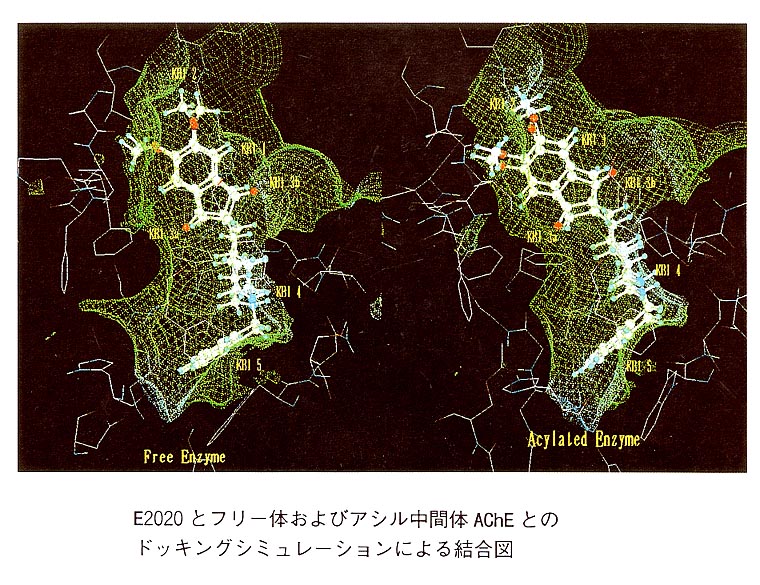
〔アセチルコリンエステラーゼ阻害剤E2020のドッキングシミユレーション]
我々が開発したアルッハイマー病治療剤E2020(欧米商品名ARlCEPT)の研究において、構造活性相関(2)と阻害メカニズムについて報告してきた(3)。そのなかで分子設計に用いた活性構造仮説が正しいか検証する目的でアセチルコリンエステラーゼ(AChE)とE2020およびその誘導体のドッキングシミュレーションを行った(4)。
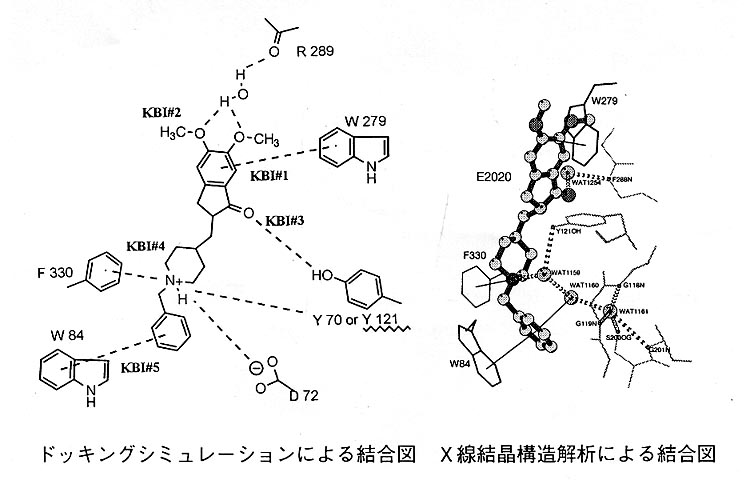
その結果、1)酵素活性部位に結合しないこと、2)複数の安定な相互作用様式が可能であること、3)AChEのアシル中間体、フリー体両方に結合が可能であることがわかった。少し詳しく説明すると、E2020はその構造中5つの部分とAChEとの間にKey
Binding Interaction (KBI)
昨年、ワイツマン研究所のSussmanらがE2020とAChEとの分解能2.5Aの複合体X線結晶構造解析に成功した(5)。その結果は我々の予想した結合様式と概ね一致していたが、必須構造であるインダノン環カルボニル基と結合していたのは水分子であった。
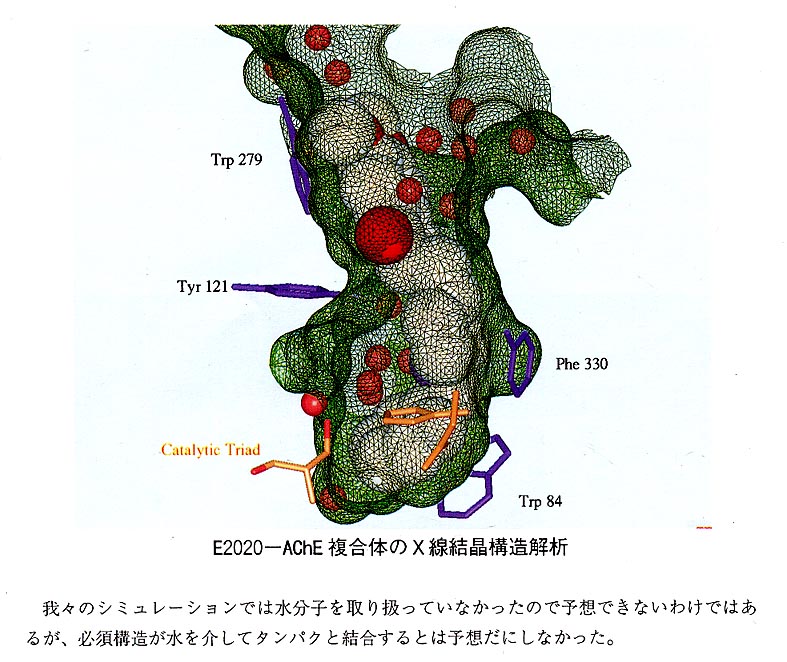
我々のシミュレーションでは水分子を取り扱っていなかったので予想できないわけではあるが、必須構造が水を介してタンパクと結合するとは予想だにしなかった。
SBDDの課題
CADDにおけるシミュレーションの正確さのレベルはまだ低く、例えば薬物と標的タンパク間の相互作用エネルギーの見積については活性との相関がないなど、構造活性相関がとれない問題を抱えている。この問題は、結合した時の相互作用エネルギーの見積の確度が低いことと、結合過程の自由エネルギーの見積ができないことなどから生じるものであろう。
今後のシミュレーション法の改良については、水の取り扱いやエントロピー項導入などさらなる計算化学の進歩を必要とする。我々も独自に水を含んだ新しいシミュレーション法を開発中であるが、今後、量子化学を取り込んだ分子動力学計算法などが期待できる。
また、著者らはタンパク質の構造解析の経験が浅く、標的タンパク作製から結晶化までのスピードが遅く、しかも標的タンパクの供給が十分でない。それゆえ、探索研究でのスピードについて行けない。これは何も我が社の構造生物学アプローチに限ったことではないと思うが、なんとかスピードをあげる工夫をしたい。
リード化合物の種になるライブラリーについては、CADDで用いる三次元データベースに今までに報告されている活性化合物とか、コンビケムで合成できそうな化合物の仮想ライブラリーを加えるなど、化合物の構造の範囲を広げることを考えていくべきである。そうしてCADDとコンビケムを掛け合わせることによりリード化合物発見の確率を高められるようになるだろう。
おわりに
創薬は、いろいろな分野の科学者によって行われる組織だった努力の成果である。以前、薬は合成と評価の繰り返しによる医薬品化学の標準的な方法で、化学前駆体を系統的に修飾することにより設計されていた。標的部位の詳細な構造に基づき、より理論的な方法で薬物設計できることを多くのメディシナルケミストは夢見てきた。
この夢は標的分子と化合物が相互作用している三次元構造を視覚的に捕らえるという構造生物学に基づいた技術により可能となった。リガンドのコンフォメーションや標的分子との相互作用に基づき、薬としての性能を高めた新しい化学物質を設計することができる。この方法ではタンパク質一阻害剤の結合様式を確認しながら、より親和性が高くなるように阻害剤の修飾、改変を行なっていくので、一旦このような系が確立されれば非常に効率の良い薬物設計が可能になる。従来、製薬企業に於いて、このようなアプローチが敬遠されがちで った原因の一つは、たとえばタンパク質のX線結晶構造解析に時間がかかり過ぎる点にあった。しかし、遺伝子工学技術の進歩により目的のタンパク質が比較的容易に得られるようになったことや、タンパク質の結晶化について過去の経験がかなり蓄積され、結晶化条件の検索が迅速になりつつあること、また、放射光の利用も含めたX線回折データ測定装置及び計算機の進歩、そして、解析技術の進歩とソフトウエアの整備などの要因により、構造解析に要する時間が大きく削減され、薬物開発のスピードに十分間に合うようぁw)ノなりつつある現在、状況はかなり変化したと言える。つまり、放射光、ロボット、高磁場、高速計算チップ、グラフィックスツールなど構造科学の分野における技術開発は、標的分子の三次元構造決定効率化における障害を著しく減らすことにより大きく改善してきた。
それらの技術は、薬物設計者が標的分子の薬物結合部位での薬物との相互作用を視覚的に捉えることを可能にし、詳細に分析してその相互作用を形成する力を評価する手段となる。視覚化と分析により一連の化合物群における結合の違いや、結合の結果生じた標的分子のコンフォメーション変化を知ることが出来る。
薬物分子と標的分子それぞれの三次元構造の役割を理解する事は、多くの疾病原因分子の機能性を新たに見い出すもととなる。さらに、CADD(Computer-aided
Drug Design)とコンビケム(Combinatorial
Chemistry)を掛け合わせる事により創薬における新しい道が開けてくるだろう。
参考文献
1 Boyd, D.B., Modern Drug Discovery, 1998, 1, 41.
2 a) Sugimoto, H.; Tsuchiya, Y.; Sugumi, H.; Higurashi, K.; Karibe,
N.; Iimura, Y.; Sasaki, A; Kawakami, Y.; Nakamura, T.; Araki, S.;
Yamanishi, Y.; Yamatsu, K. J. Med.
Chem., 1990, 33, 1880.
b) Sugimoto, H.; Tsuchiya, Y.; Sugumi, H.; Higurashi,
K.; Karibe, N.; Iimura, Y.; Sasaki, A.; Araki, S.; Yamanishi, Y.; Yamatsu,
K. J. Med.
Chem., 1992, 35,
4542.
c) Sugimoto, H.; Iimura, Y.; Yamanishi, Y.; Yamatsu, K.
J. Med. Chem., 1995, 38, 4821.
d) Cardozo, M.; Kawai, T.; Iimura, Y.; Sugimoto, H.;
Yamanishi,
Y.; Hopfinger, A. J. Med. Chem., 1992, 35, 590.
e) Cardozo, M.G.; Iimura, Y.; Sugimoto, H.; Yamanishi,
Y.; Hopfinger, A.J. J. Med. Chem., 1992, 35. 584.
3 Nochi,S.; Asakawa, N.; Sato, T. Biol. Pharm. Bull.,
1995,
18, 1145.
4 a) Inoue, A.; Kawai,T.; Wakita, M.; Iimura, Y.; Sugimoto, H.;
Kawakami,
Y. J. Med. Chem., 1996, 39, 4460.
b) Kawakami, Y.; Inoue, A.; Kawai, T.; Wakita, M.;
Sugimoto,
H.; Hopfinger, A. BioOrg. Med. Chem.. 1996, 4, 1429.
5 a) Kryger, G.; Silman, I.; Sussman, J.L. J. Physiol. Paris,
1998, 92, 191.
b) Kryger, G.; Silman, I.; Sussman, J.L. Structure,
1999, 7, 297.